DNase footprint profiles using ENCODE DNase-seq data
Kaixuan Luo
7/31/2018
Last updated: 2018-08-29
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(20180613)The command
set.seed(20180613)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 2f894eb
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: code_RCC/.DS_Store Ignored: data/.DS_Store Untracked files: Untracked: analysis/ATACseq_footprint_profiles_GGR_Duke.Rmd Untracked: analysis/ATACseq_footprint_profiles_OliviaGray.Rmd Untracked: analysis/ATACseq_footprint_profiles_OliviaGray_HAEC.Rmd Untracked: analysis/ATACseq_preprocessing_pipeline_GGR_hg38.Rmd Untracked: code_RCC/update_bamnames.R Untracked: docs/figure/ATACseq_footprint_profiles_GGR_Duke.Rmd/ Untracked: docs/figure/ATACseq_footprint_profiles_OliviaGray.Rmd/ Untracked: docs/figure/ATACseq_footprint_profiles_OliviaGray_HAEC.Rmd/ Untracked: docs/figure/ATACseq_footprint_profiles_smooth.Rmd/ Untracked: workflow_setup.R Unstaged changes: Modified: analysis/ATACseq_preprocessing_pipeline.Rmd Modified: code_RCC/fimo_jaspar_motif_rcc.sh Modified: code_RCC/genome_coverage_bamToBigwig_GGR_hg38.sh Modified: code_RCC/get_motif_count_matrices_GGR_hg38.sh
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 2f894eb | kevinlkx | 2018-08-29 | plot DNase footprint profiles of CTCF and REST using ENCODE data |
| html | 0e20593 | kevinlkx | 2018-08-29 | Build site. |
| Rmd | a233f11 | kevinlkx | 2018-08-29 | plot DNase footprint profiles of CTCF and REST using ENCODE data |
| html | b182c05 | kevinlkx | 2018-08-29 | Build site. |
| Rmd | 09836f9 | kevinlkx | 2018-08-29 | plot DNase footprint profiles of CTCF and REST using ENCODE data |
| html | 35c5feb | kevinlkx | 2018-08-29 | Build site. |
| Rmd | f318dd5 | kevinlkx | 2018-08-29 | plot DNase footprint profiles of CTCF and REST using ENCODE data |
functions
##### Functions #####
## load and combine count matrices
load_combine_counts <- function(tf_name, pwm_name, dir_count_matrix){
cat("Loading count matrices ... \n")
counts_fwd.df <- read.table(paste0(dir_count_matrix, "/", tf_name, "/", pwm_name, "_hg19_dnase_fwdcounts.m.gz"))
counts_rev.df <- read.table(paste0(dir_count_matrix, "/", tf_name, "/", pwm_name, "_hg19_dnase_revcounts.m.gz"))
## the first 5 columns from "bwtool extract" are chr, start, end, name, and the number of data points
counts_fwd.df <- counts_fwd.df[, -c(1:5)]
counts_rev.df <- counts_rev.df[, -c(1:5)]
colnames(counts_fwd.df) <- paste0("fwd", 1:ncol(counts_fwd.df))
colnames(counts_rev.df) <- paste0("rev", 1:ncol(counts_rev.df))
counts_combined.m <- as.matrix(cbind(counts_fwd.df, counts_rev.df))
return(counts_combined.m)
}
## select candidate sites by mapability and PWM score cutoffs
select_sites <- function(sites.df, thresh_mapability=NULL, thresh_PWMscore=NULL, readstats_name=NULL){
# cat("loading sites ...\n")
cat("Select candidate sites \n")
if(!is.null(thresh_mapability) || !is.na(thresh_mapability)){
cat("Select candidate sites with mapability >=", thresh_mapability, "\n")
idx_mapability <- (sites.df[,"mapability"] >= thresh_mapability)
}else{
idx_mapability <- rep(TRUE, nrow(sites.df))
}
if(!is.null(thresh_PWMscore) || !is.na(thresh_PWMscore)){
cat("Select candidate sites with PWM score >=", thresh_PWMscore, "\n")
idx_pwm <- (sites.df[,"pwm_score"] >= thresh_PWMscore)
}else{
idx_pwm <- rep(TRUE, nrow(sites.df))
}
if(!is.null(readstats_name)){
readstats.df <- read.table(readstats_name, header = F)
## if the readstats.df contains chrY, then it means the cell type is male, then the candidate sites should contain chrY,
## otherwise, the cell type is female, then the candidate sites on chrY should be removed.
if( "chrY" %in% readstats.df[,1] ){
cat("include chrY sites \n")
idx_chr <- (sites.df[,1] != "")
}else{
cat("chrY NOT in the bam file, filter out chrY sites \n")
## remove chrY from candidate (motif) sites
idx_chr <- (sites.df[,1] != "chrY")
}
}else{
idx_chr <- rep(TRUE, nrow(sites.df))
}
idx_select <- which(idx_mapability & idx_pwm & idx_chr)
return(idx_select)
}parameters
ver_genome <- "hg19"
flank <- 100
thresh_mapability <- 0.8
thresh_PWMscore <- 10
num_top_sites <- 1000 # plot top sites
max_cuts <- 20 # Clip extreme values
dir_data <- "~/Dropbox/research/ATAC_DNase/"CTCF in GM12878 cell type
load DNase footprint data
cell_type <- "GM12878"
tf_name <- "CTCF"
pwm_name <- "CTCF_MA0139.1_1e-5"
dir_count_matrix <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/")
dir_sites_chip <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/ChIPSeq/")
filename_sites <- paste0(dir_sites_chip, "/", "chipseq_", cell_type, "_", pwm_name, "_flank", flank, "_exp1.totalcount")
sites.df <- read.table(filename_sites, header = T, comment.char = "!", stringsAsFactors = F)
sites.df <- sites.df[, c("chr", "start", "end", "site", "pwmScore", "strand", "pValue", "mapability", "ChIP_mean")]
colnames(sites.df) = c("chr", "start", "end", "name", "pwm_score", "strand", "p_value", "mapability", "ChIP")
idx_select <- select_sites(sites.df, thresh_mapability, thresh_PWMscore)Select candidate sites
Select candidate sites with mapability >= 0.8
Select candidate sites with PWM score >= 10 sites.df <- sites.df[idx_select, ]
cat("Number of sites:", nrow(sites.df), "\n")Number of sites: 54859 counts_combined.m <- load_combine_counts(tf_name, pwm_name, dir_count_matrix)Loading count matrices ... counts_combined.m <- counts_combined.m[idx_select,]
## Clip extreme values
counts_combined.m[counts_combined.m > max_cuts] <- max_cuts
cat("Dimension of", dim(counts_combined.m), "\n")Dimension of 54859 436 if(nrow(counts_combined.m) != nrow(sites.df)){
stop("Sites not matched!")
}plot footprint profiles of highest occupancy
order_selected <- order(sites.df$ChIP, decreasing = T)[1:num_top_sites]
counts_selected.m <- counts_combined.m[order_selected,]
counts_profile <- apply(counts_selected.m, 2, mean)
par(mfrow = c(1,2))
counts <- counts_profile[1:(length(counts_profile)/2)]
plot(counts, type = "l", col = "blue", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "forward strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)
counts <- counts_profile[(length(counts_profile)/2+1): length(counts_profile)]
plot(counts, type = "l", col = "red", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "reverse strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)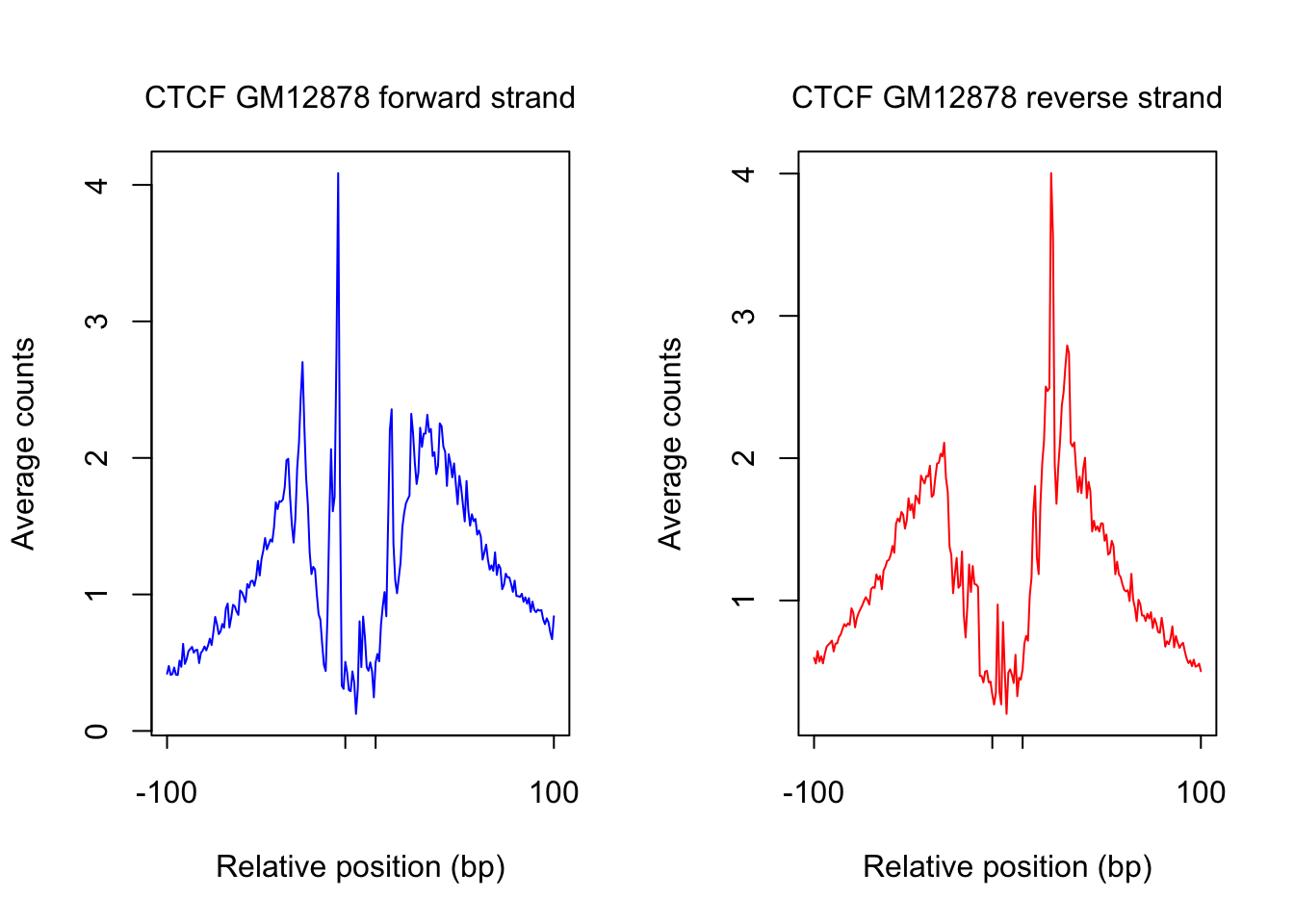
Expand here to see past versions of unnamed-chunk-4-1.png:
| Version | Author | Date |
|---|---|---|
| 35c5feb | kevinlkx | 2018-08-29 |
## save counts matrix
rds_name <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/", tf_name, "/",
pwm_name, "_hg19_dnase_fwdcounts_selected_sites.rds")
saveRDS(counts_selected.m, rds_name)plot footprint profiles of most accessible sites
order_selected <- order(rowSums(counts_combined.m), decreasing = T)[1:num_top_sites]
counts_selected.m <- counts_combined.m[order_selected,]
counts_profile <- apply(counts_selected.m, 2, mean)
par(mfrow = c(1,2))
counts <- counts_profile[1:(length(counts_profile)/2)]
plot(counts, type = "l", col = "blue", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "forward strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)
counts <- counts_profile[(length(counts_profile)/2+1): length(counts_profile)]
plot(counts, type = "l", col = "red", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "reverse strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)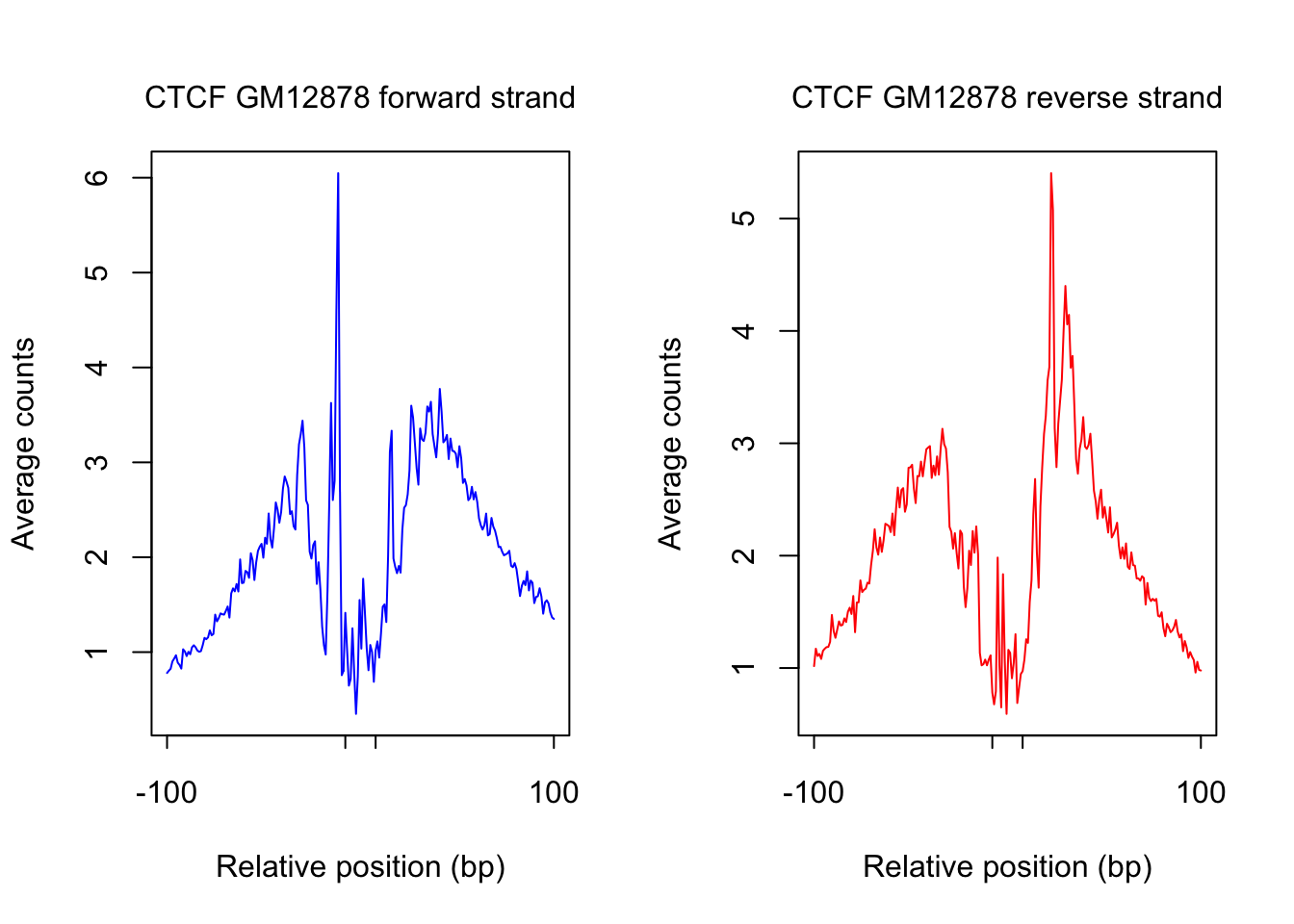
Expand here to see past versions of unnamed-chunk-5-1.png:
| Version | Author | Date |
|---|---|---|
| 35c5feb | kevinlkx | 2018-08-29 |
CTCF in K562 cell type
load DNase footprint data
cell_type <- "K562"
tf_name <- "CTCF"
pwm_name <- "CTCF_MA0139.1_1e-5"
dir_count_matrix <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/")
dir_sites_chip <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/ChIPSeq/")
filename_sites <- paste0(dir_sites_chip, "/", "chipseq_", cell_type, "_", pwm_name, "_flank", flank, "_exp1.totalcount")
sites.df <- read.table(filename_sites, header = T, comment.char = "!", stringsAsFactors = F)
sites.df <- sites.df[, c("chr", "start", "end", "site", "pwmScore", "strand", "pValue", "mapability", "ChIP_mean")]
colnames(sites.df) = c("chr", "start", "end", "name", "pwm_score", "strand", "p_value", "mapability", "ChIP")
idx_select <- select_sites(sites.df, thresh_mapability, thresh_PWMscore)Select candidate sites
Select candidate sites with mapability >= 0.8
Select candidate sites with PWM score >= 10 sites.df <- sites.df[idx_select, ]
cat("Number of sites:", nrow(sites.df), "\n")Number of sites: 54859 counts_combined.m <- load_combine_counts(tf_name, pwm_name, dir_count_matrix)Loading count matrices ... counts_combined.m <- counts_combined.m[idx_select,]
## Clip extreme values
counts_combined.m[counts_combined.m > max_cuts] <- max_cuts
cat("Dimension of", dim(counts_combined.m), "\n")Dimension of 54859 436 if(nrow(counts_combined.m) != nrow(sites.df)){
stop("Sites not matched!")
}plot footprint profiles of highest occupancy sites
order_selected <- order(sites.df$ChIP, decreasing = T)[1:num_top_sites]
counts_selected.m <- counts_combined.m[order_selected,]
counts_profile <- apply(counts_selected.m, 2, mean)
par(mfrow = c(1,2))
counts <- counts_profile[1:(length(counts_profile)/2)]
plot(counts, type = "l", col = "blue", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "forward strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)
counts <- counts_profile[(length(counts_profile)/2+1): length(counts_profile)]
plot(counts, type = "l", col = "red", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "reverse strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)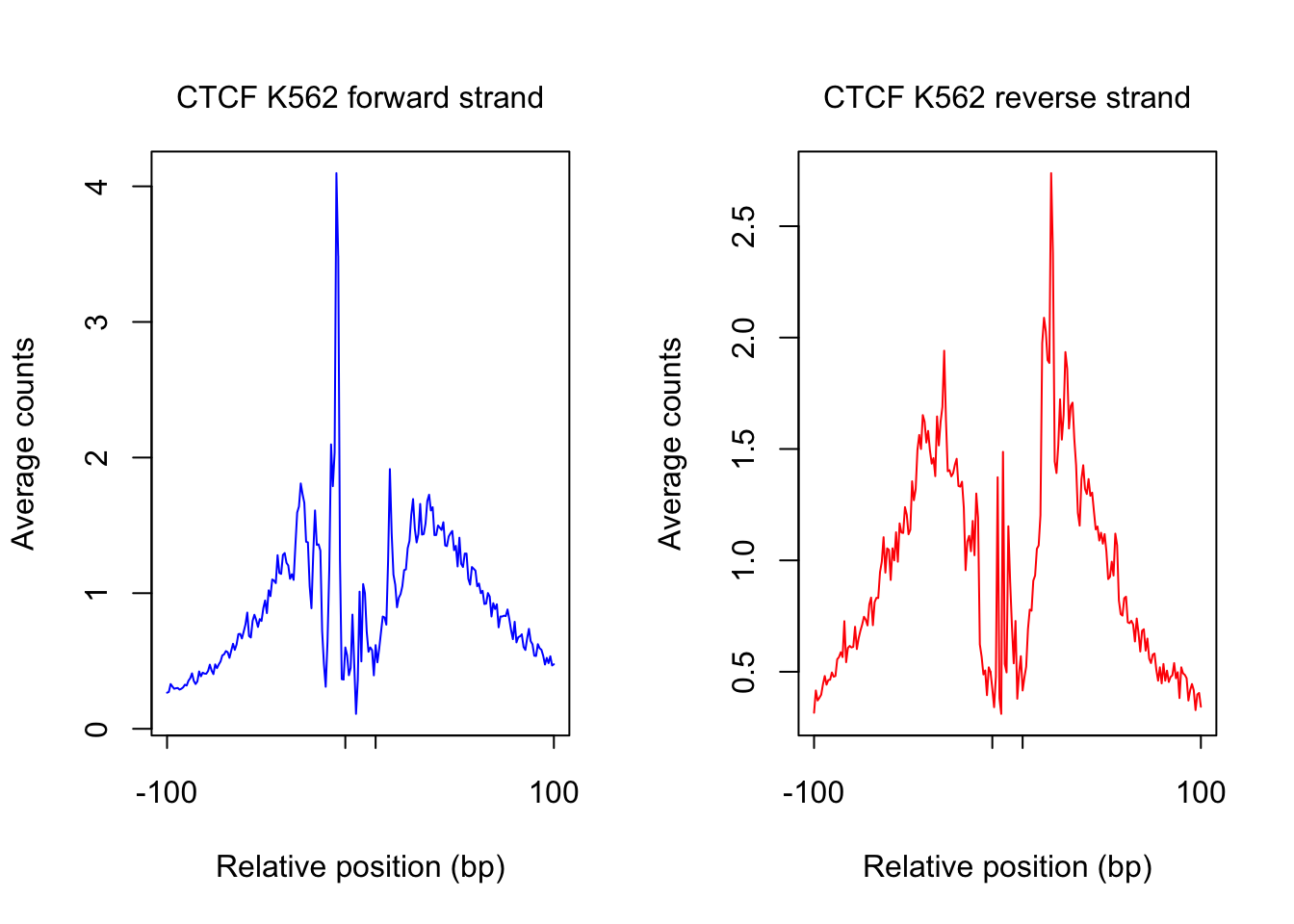
Expand here to see past versions of unnamed-chunk-7-1.png:
| Version | Author | Date |
|---|---|---|
| 35c5feb | kevinlkx | 2018-08-29 |
## save counts matrix
rds_name <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/", tf_name, "/",
pwm_name, "_hg19_dnase_fwdcounts_selected_sites.rds")
saveRDS(counts_selected.m, rds_name)REST in GM12878 cell type
load DNase footprint data
cell_type <- "GM12878"
tf_name <- "REST"
pwm_name <- "REST_MA0138.2_1e-5"
dir_count_matrix <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/")
dir_sites_chip <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/ChIPSeq/")
filename_sites <- paste0(dir_sites_chip, "/", "chipseq_", cell_type, "_", pwm_name, "_flank", flank, "_exp1.totalcount")
sites.df <- read.table(filename_sites, header = T, comment.char = "!", stringsAsFactors = F)
sites.df <- sites.df[, c("chr", "start", "end", "site", "pwmScore", "strand", "pValue", "mapability", "ChIP_mean")]
colnames(sites.df) = c("chr", "start", "end", "name", "pwm_score", "strand", "p_value", "mapability", "ChIP")
idx_select <- select_sites(sites.df, thresh_mapability, thresh_PWMscore)Select candidate sites
Select candidate sites with mapability >= 0.8
Select candidate sites with PWM score >= 10 sites.df <- sites.df[idx_select, ]
cat("Number of sites:", nrow(sites.df), "\n")Number of sites: 54533 counts_combined.m <- load_combine_counts(tf_name, pwm_name, dir_count_matrix)Loading count matrices ... counts_combined.m <- counts_combined.m[idx_select,]
## Clip extreme values
counts_combined.m[counts_combined.m > max_cuts] <- max_cuts
cat("Dimension of", dim(counts_combined.m), "\n")Dimension of 54533 440 if(nrow(counts_combined.m) != nrow(sites.df)){
stop("Sites not matched!")
}plot footprint profiles of highest occupancy sites
order_selected <- order(sites.df$ChIP, decreasing = T)[1:num_top_sites]
counts_selected.m <- counts_combined.m[order_selected,]
counts_profile <- apply(counts_selected.m, 2, mean)
par(mfrow = c(1,2))
counts <- counts_profile[1:(length(counts_profile)/2)]
plot(counts, type = "l", col = "blue", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "forward strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)
counts <- counts_profile[(length(counts_profile)/2+1): length(counts_profile)]
plot(counts, type = "l", col = "red", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "reverse strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)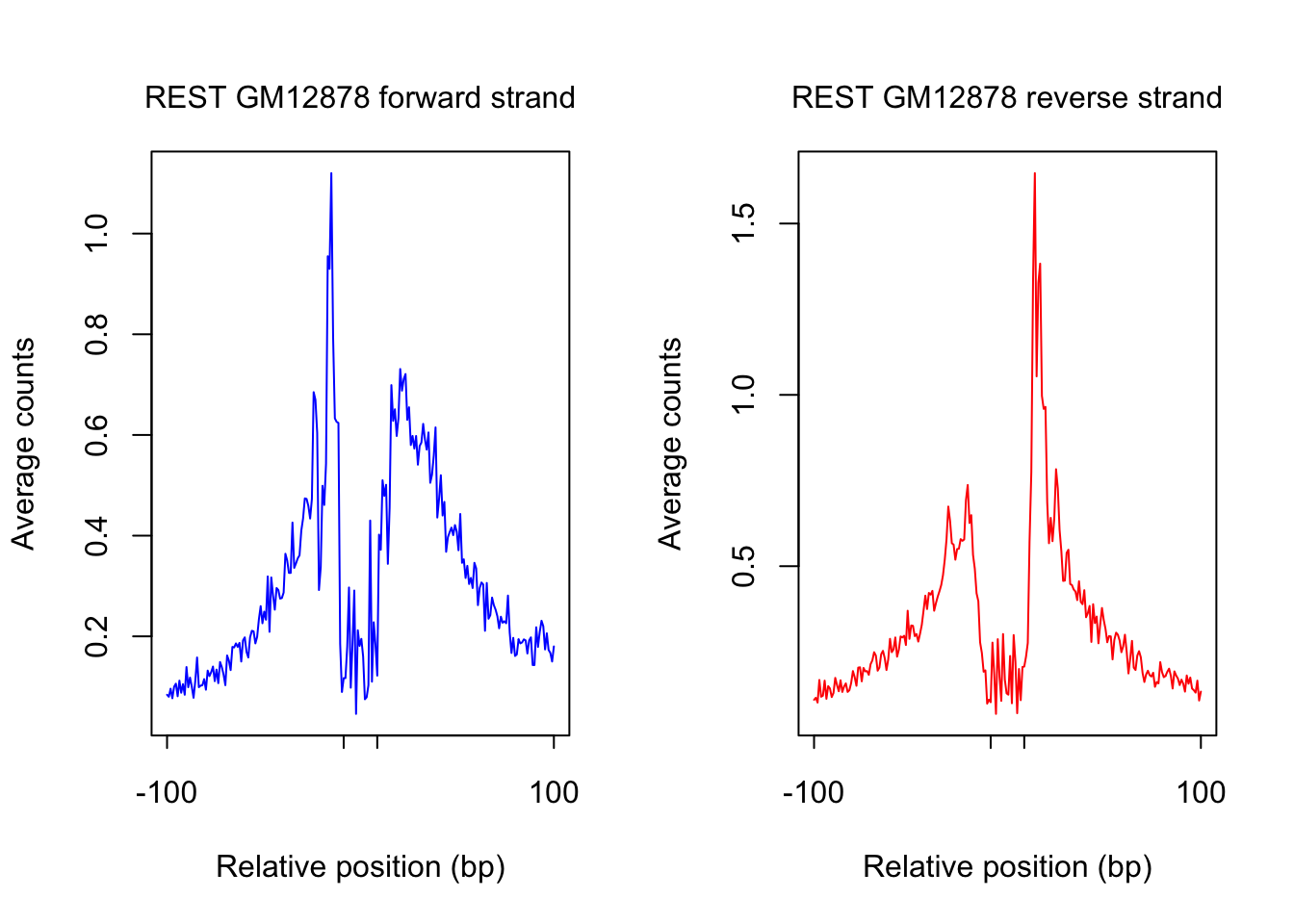
Expand here to see past versions of unnamed-chunk-9-1.png:
| Version | Author | Date |
|---|---|---|
| 0e20593 | kevinlkx | 2018-08-29 |
## save counts matrix
rds_name <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/", tf_name, "/",
pwm_name, "_hg19_dnase_fwdcounts_selected_sites.rds")
saveRDS(counts_selected.m, rds_name)plot footprint profiles of most accessible sites
order_selected <- order(rowSums(counts_combined.m), decreasing = T)[1:num_top_sites]
counts_selected.m <- counts_combined.m[order_selected,]
counts_profile <- apply(counts_selected.m, 2, mean)
par(mfrow = c(1,2))
counts <- counts_profile[1:(length(counts_profile)/2)]
plot(counts, type = "l", col = "blue", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "forward strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)
counts <- counts_profile[(length(counts_profile)/2+1): length(counts_profile)]
plot(counts, type = "l", col = "red", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "reverse strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)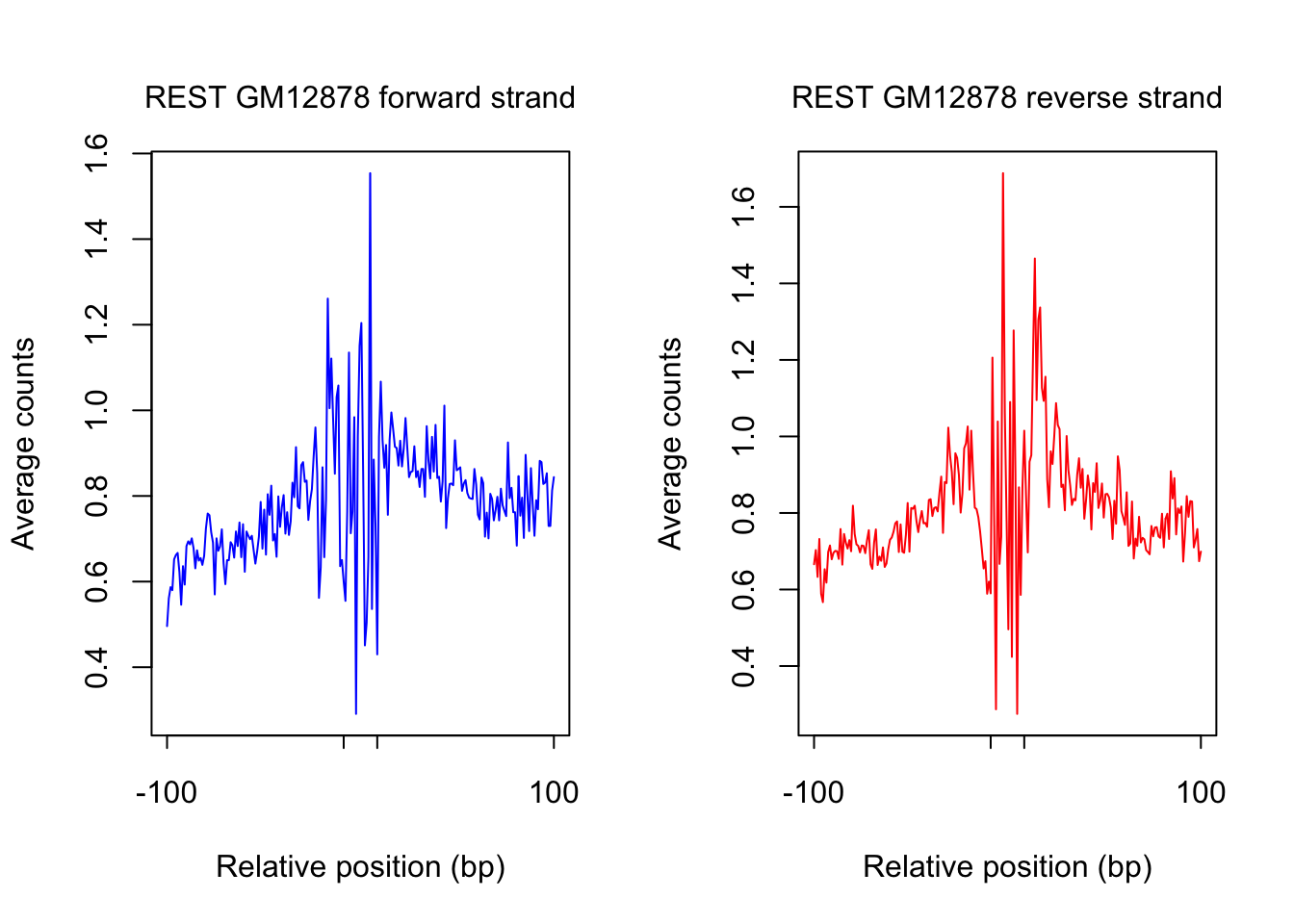
REST in K562 cell type
load DNase footprint data
cell_type <- "K562"
tf_name <- "REST"
pwm_name <- "REST_MA0138.2_1e-5"
dir_count_matrix <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/")
dir_sites_chip <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/ChIPSeq/")
filename_sites <- paste0(dir_sites_chip, "/", "chipseq_", cell_type, "_", pwm_name, "_flank", flank, "_exp1.totalcount")
sites.df <- read.table(filename_sites, header = T, comment.char = "!", stringsAsFactors = F)
sites.df <- sites.df[, c("chr", "start", "end", "site", "pwmScore", "strand", "pValue", "mapability", "ChIP_mean")]
colnames(sites.df) = c("chr", "start", "end", "name", "pwm_score", "strand", "p_value", "mapability", "ChIP")
idx_select <- select_sites(sites.df, thresh_mapability, thresh_PWMscore)Select candidate sites
Select candidate sites with mapability >= 0.8
Select candidate sites with PWM score >= 10 sites.df <- sites.df[idx_select, ]
cat("Number of sites:", nrow(sites.df), "\n")Number of sites: 54533 counts_combined.m <- load_combine_counts(tf_name, pwm_name, dir_count_matrix)Loading count matrices ... counts_combined.m <- counts_combined.m[idx_select,]
## Clip extreme values
counts_combined.m[counts_combined.m > max_cuts] <- max_cuts
cat("Dimension of", dim(counts_combined.m), "\n")Dimension of 54533 440 if(nrow(counts_combined.m) != nrow(sites.df)){
stop("Sites not matched!")
}plot footprint profiles of highest occupancy sites
order_selected <- order(sites.df$ChIP, decreasing = T)[1:num_top_sites]
counts_selected.m <- counts_combined.m[order_selected,]
counts_profile <- apply(counts_selected.m, 2, mean)
par(mfrow = c(1,2))
counts <- counts_profile[1:(length(counts_profile)/2)]
plot(counts, type = "l", col = "blue", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "forward strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)
counts <- counts_profile[(length(counts_profile)/2+1): length(counts_profile)]
plot(counts, type = "l", col = "red", xlab = "Relative position (bp)", ylab = "Average counts",
main = "", xaxt = "n")
mtext(text = paste(tf_name, cell_type, "reverse strand"), side = 3, line = 1, cex = 1)
axis(1,at=c(1, flank+1, length(counts)-flank, length(counts)), labels=c(-flank, '','' ,flank),
cex.axis = 1, tck=-0.03, tick = T, cex = 1)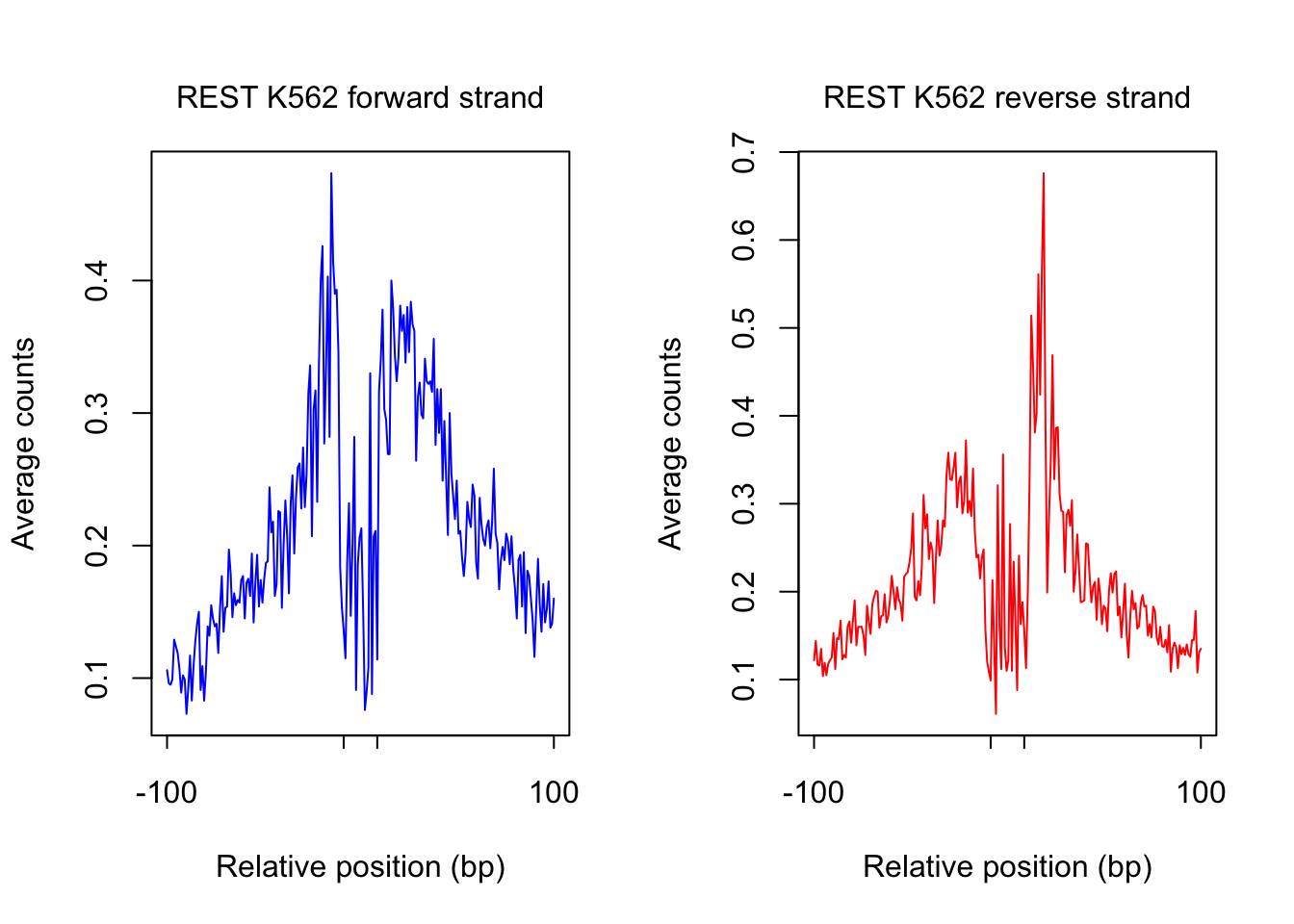
Expand here to see past versions of unnamed-chunk-12-1.png:
| Version | Author | Date |
|---|---|---|
| 0e20593 | kevinlkx | 2018-08-29 |
## save counts matrix
rds_name <- paste0(dir_data, "/DNase-seq_ENCODE/", cell_type, "/DNaseSeq/DNase_tagcount_matrix/", tf_name, "/",
pwm_name, "_hg19_dnase_fwdcounts_selected_sites.rds")
saveRDS(counts_selected.m, rds_name)Session information
sessionInfo()R version 3.4.3 (2017-11-30)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] workflowr_1.1.1 Rcpp_0.12.16 digest_0.6.15
[4] rprojroot_1.3-2 R.methodsS3_1.7.1 backports_1.1.2
[7] git2r_0.21.0 magrittr_1.5 evaluate_0.10.1
[10] stringi_1.1.7 whisker_0.3-2 R.oo_1.22.0
[13] R.utils_2.6.0 rmarkdown_1.9 tools_3.4.3
[16] stringr_1.3.0 yaml_2.1.18 compiler_3.4.3
[19] htmltools_0.3.6 knitr_1.20 This reproducible R Markdown analysis was created with workflowr 1.1.1