Training model using using trendiflter
Joyce Hsiao
Last updated: 2018-07-02
Code version: 07ea531
Packages
library(Biobase)Use the top 10 genes identified to have cyclical expression patterns, in one training dataset
Get data
df <- readRDS(file="../data/eset-final.rds")
pdata <- pData(df)
fdata <- fData(df)
# select endogeneous genes
counts <- exprs(df)[grep("ENSG", rownames(df)), ]
log2cpm.all <- t(log2(1+(10^6)*(t(counts)/pdata$molecules)))
macosko <- readRDS("../data/cellcycle-genes-previous-studies/rds/macosko-2015.rds")
log2cpm.all <- log2cpm.all[,order(pdata$theta)]
pdata <- pdata[order(pdata$theta),]
log2cpm.quant <- readRDS("../output/npreg-trendfilter-quantile.Rmd/log2cpm.quant.rds")
# import previously identifid cell cycle genes
# cyclegenes <- readRDS("../output/npreg-methods.Rmd/cyclegenes.rds")
# cyclegenes.names <- colnames(cyclegenes)[2:6]
# select external validation samples
set.seed(99)
nvalid <- round(ncol(log2cpm.quant)*.15)
ii.valid <- sample(1:ncol(log2cpm.quant), nvalid, replace = F)
ii.nonvalid <- setdiff(1:ncol(log2cpm.quant), ii.valid)
log2cpm.quant.nonvalid <- log2cpm.quant[,ii.nonvalid]
log2cpm.quant.valid <- log2cpm.quant[,ii.valid]
theta <- pdata$theta
names(theta) <- rownames(pdata)
theta.nonvalid <- theta[ii.nonvalid]
theta.valid <- theta[ii.valid]
sig.genes <- readRDS("../output/npreg-trendfilter-quantile.Rmd/out.stats.ordered.sig.rds")
expr.sig <- log2cpm.quant.nonvalid[rownames(log2cpm.quant.nonvalid) %in% rownames(sig.genes), ]
# set training samples
source("../peco/R/primes.R")
source("../peco/R/partitionSamples.R")
parts <- partitionSamples(1:ncol(log2cpm.quant.nonvalid), runs=5,
nsize.each = rep(151,5))
part_indices <- parts$partitionssource("../peco/R/fit.trendfilter.generic.R")
source("../peco/R/cycle.npreg.R")
source("../peco/R/cycle.corr.R")
Y_train <- expr.sig[,part_indices[[1]]$train]
theta_train <- theta.nonvalid[part_indices[[1]]$train]- The shape of the predicted expression levels is similar between results based on fucci-labels and reuslts based on PCA of the expression levels. Hence, no need to reverse the axis.
Y_train <- expr.sig[,part_indices[[1]]$train]
theta_train <- theta.nonvalid[part_indices[[1]]$train]
theta_train_pca <- initialize_cell_times(Y_train)fit.train.nobin <- cycle.npreg.insample(Y = Y_train,
theta = theta_train,
nbins = NULL, ncores=10)
fit.train.pca.nobin <- cycle.npreg.insample(Y = Y_train,
theta = theta_train_pca,
nbins = NULL, ncores=10)
saveRDS(fit.train.nobin, "../output/method-npreg.Rmd/fit.train.nobin.rds")
saveRDS(fit.train.pca.nobin, "../output/method-npreg.Rmd/fit.train.pca.nobin.rds")fit.train.nobin <- readRDS("../output/method-npreg.Rmd/fit.train.nobin.rds")
fit.train.pca.nobin <- readRDS("../output/method-npreg.Rmd/fit.train.pca.nobin.rds")
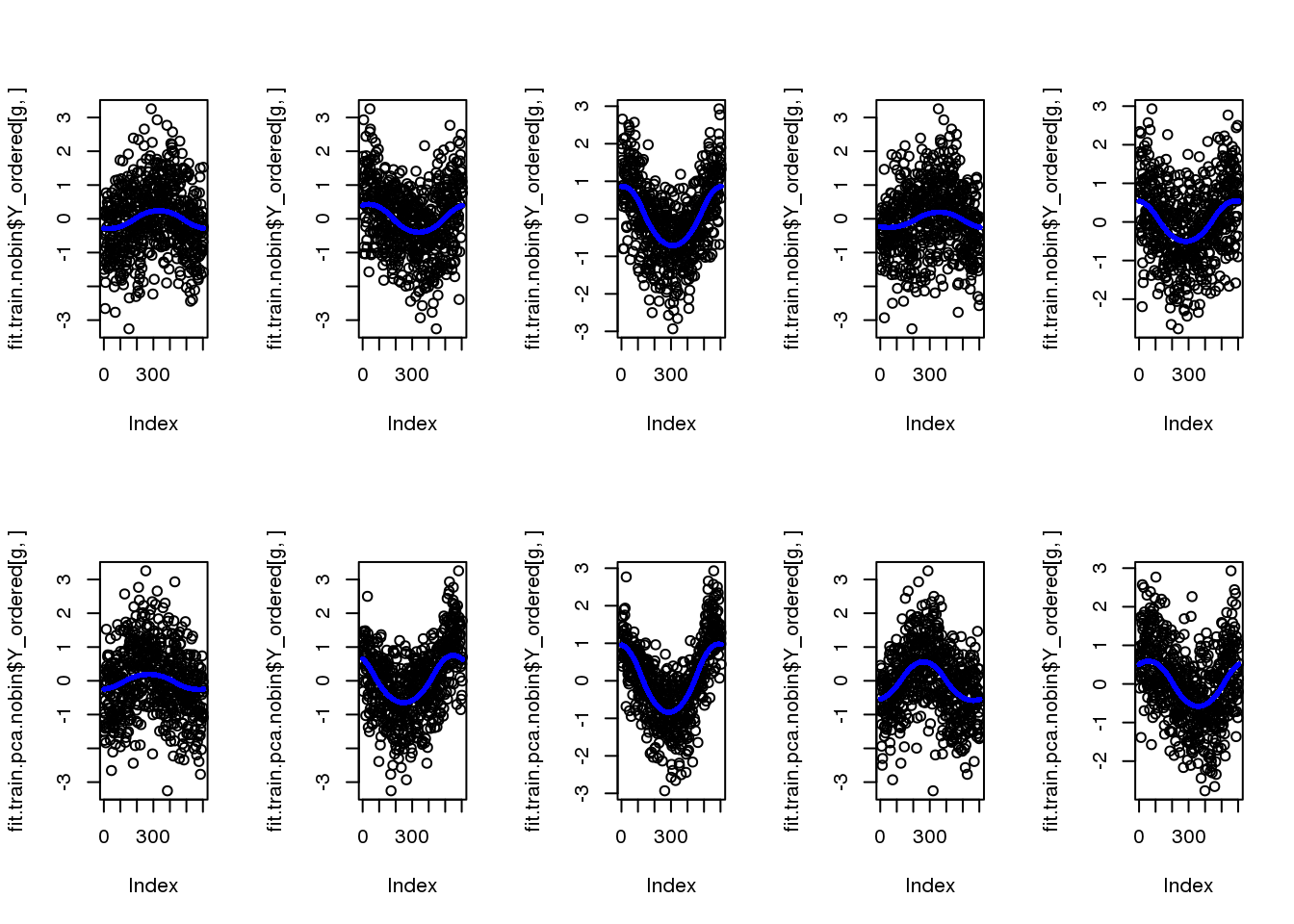
fit.train.nobin$loglik_est[1] -7578.007fit.train.pca.nobin$loglik_est[1] -6737.267par(mfcol=c(2,5))
for (g in 1:5) {
plot(fit.train.nobin$Y_ordered[g,])
points(fit.train.nobin$mu_est[g,], col="blue", cex=.6, pch=16)
plot(fit.train.pca.nobin$Y_ordered[g,])
points(fit.train.pca.nobin$mu_est[g,], col="blue", cex=.6, pch=16)
}
Fisher-Lee correlation coefficient for rotational dependence supports a significant rotational dependency between the cell times re-estimated based on FUCCI and the cell times re-estimated based on PCA.
par(mfrow=c(1,1))
plot(fit.train.nobin$cell_times_est,
fit.train.pca.nobin$cell_times_est[match(names(fit.train.nobin$cell_times_est),
names(fit.train.pca.nobin$cell_times_est))])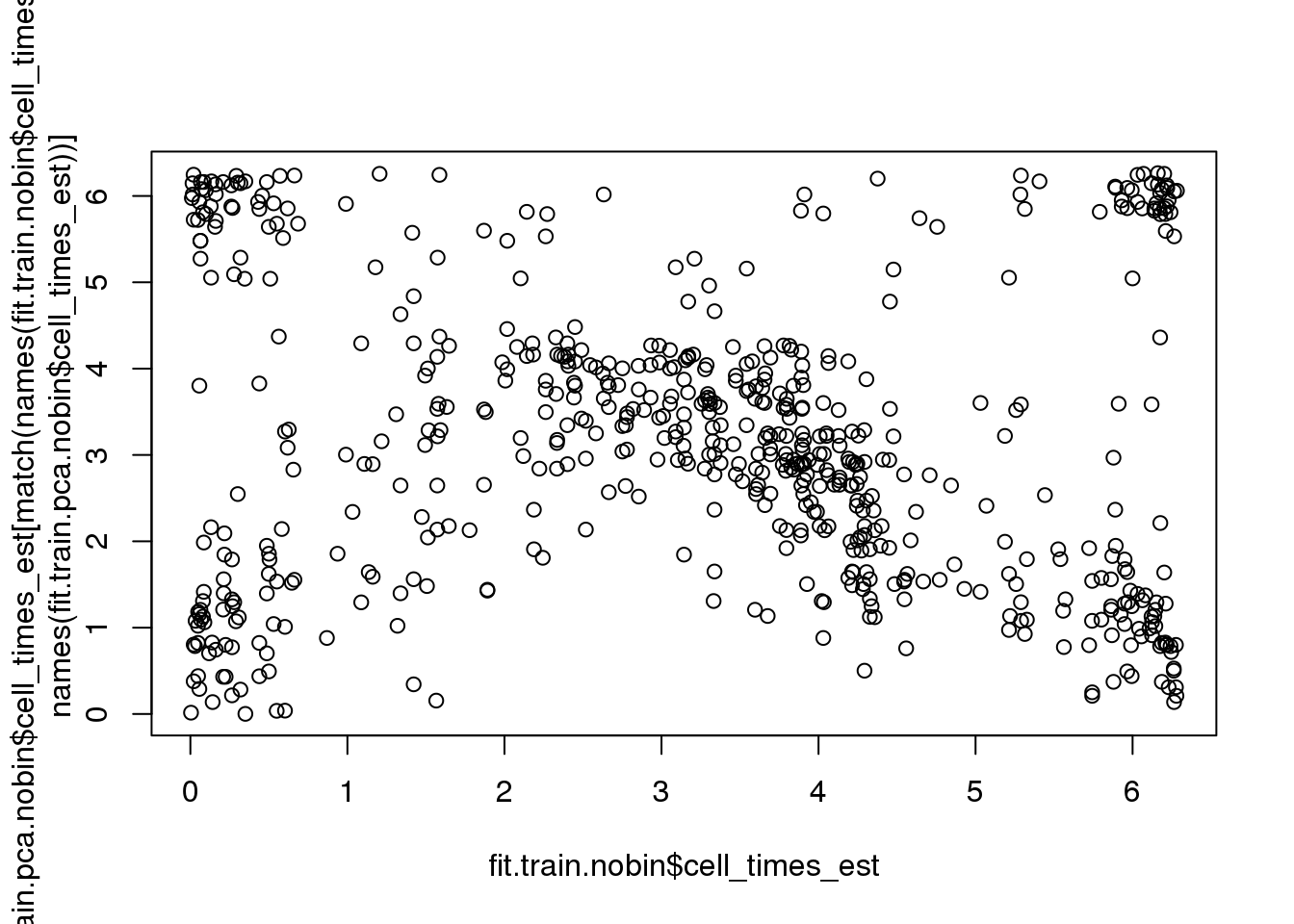
rtest_pval <- rFL.IndTestRand(fit.train.nobin$cell_times_est,
fit.train.pca.nobin$cell_times_est[match(names(fit.train.nobin$cell_times_est),
names(fit.train.pca.nobin$cell_times_est))],
NR=9999)
# rtest_boot <- rhoFLCIBoot(fit.train.nobin$cell_times_est,
# fit.train.pca.nobin$cell_times_est[match(names(fit.train.nobin$cell_times_est),
# names(fit.train.pca.nobin$cell_times_est))],
# ConfLevel = 95, B=9999)
#
# rtest_js <- JSTestRand(fit.train.nobin$cell_times_est,
# fit.train.pca.nobin$cell_times_est[match(names(fit.train.nobin$cell_times_est),
# names(fit.train.pca.nobin$cell_times_est))],
# NR=9999)
rtest_pval[1] -0.24333 0.00010Fisher-Lee correlation coefficient for rotational dependence supports a significant rotational dependency between the FUCCI cell times and cell times re-estimated based on FUCCI.
par(mfrow=c(1,1))
plot(fit.train.nobin$cell_times_est,
theta_train[match(names(fit.train.nobin$cell_times_est),
names(theta_train))])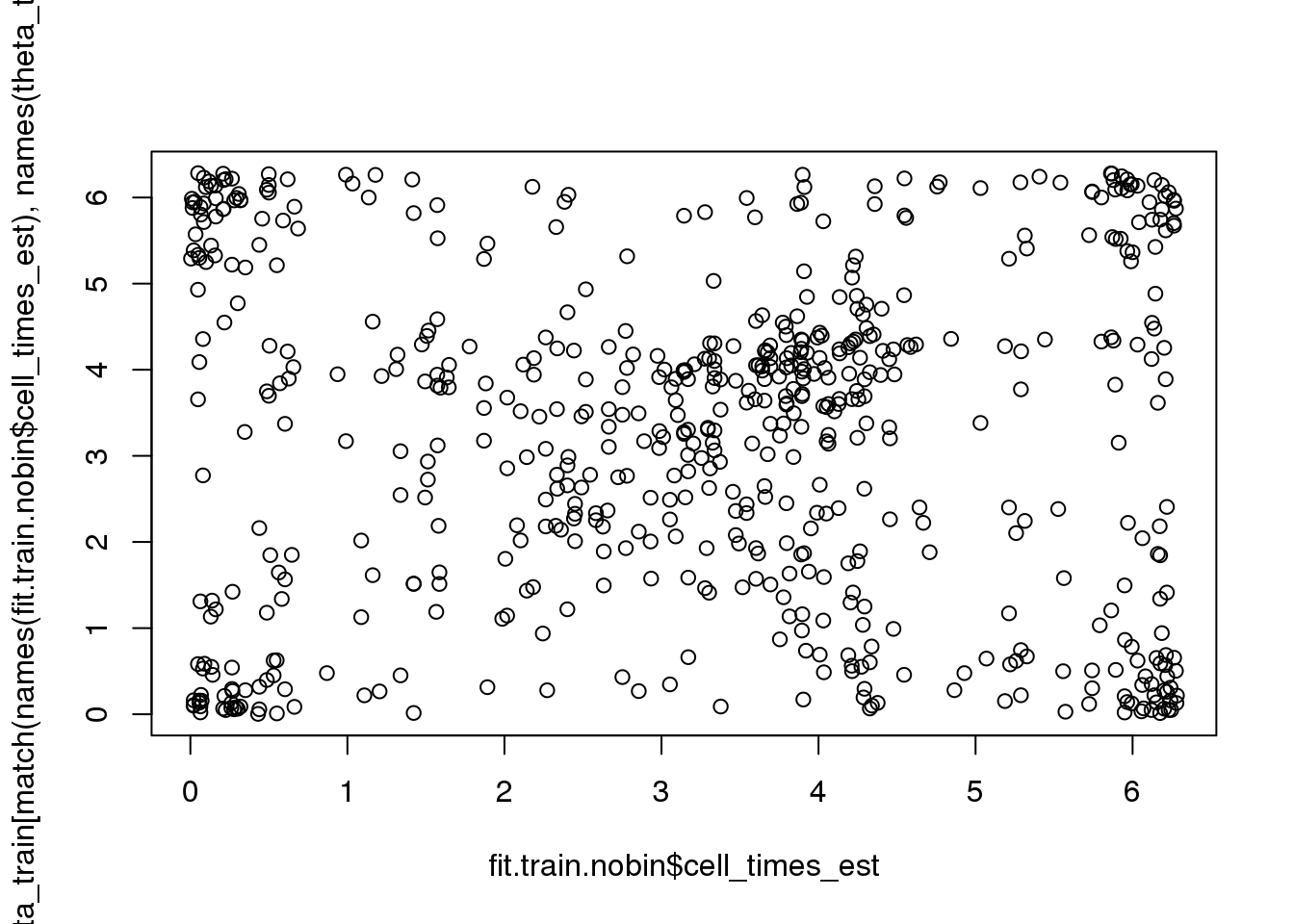
rtest_pval <- rFL.IndTestRand(fit.train.nobin$cell_times_est,
theta_train[match(names(fit.train.nobin$cell_times_est),
names(theta_train))],
NR=9999)
# rtest_boot <- rhoFLCIBoot(fit.train.nobin$cell_times_est,
# theta_train[match(names(fit.train.nobin$cell_times_est),
# names(theta_train))],
# ConfLevel = 95, B=9999)
# # a version in the circular package
# rtest_js <- JSTestRand(fit.train.nobin$cell_times_est,
# theta_train[match(names(fit.train.nobin$cell_times_est),
# names(theta_train))], NR=9999)
rtest_pval[1] 0.07064764 0.00010000# rtest_boot
# rtest_jsConsider the above fitting applying to the test sample
Compare for test samples, the ablity to predict cell times 1) using cyclical patterns for each gene predicted from the training samples initialized by fucci-labels, 2) using cyclical patterns from each gene predicted from training samples initialized by PCA, 3) using the cyclial patterns for each gene in the test samples initialized by fucc-labels, 4) using the cyclial patterns for each gene in the test samples initialized by PCA.
Y_test <- expr.sig[,part_indices[[1]]$test]
theta_test <- theta.nonvalid[part_indices[[1]]$test]
#theta_test_pca <- initialize_cell_times(Y_test)fit.test.bytrain.fucci.bin <- vector("list", 5)
for (i in 1:5) {
fit.test.bytrain.fucci.bin[[i]] <- cycle.npreg.outsample(Y_test,
theta_est=fit.train.bin$cell_times_est,
mu_est=fit.train.bin$mu_est,
sigma_est=fit.train.bin$sigma_est)
}
saveRDS(fit.test.bytrain.fucci.bin,
"../output/method-npreg.Rmd/fit.test.bytrain.fucci.bin.rds")## not specifying bins when predicting
fit.test.bytrain.fucci.nobin <- cycle.npreg.outsample(Y_test,
theta_est=fit.train.nobin$cell_times_est,
mu_est=fit.train.nobin$mu_est,
sigma_est=fit.train.nobin$sigma_est)
saveRDS(fit.test.bytrain.fucci,
"../output/method-npreg.Rmd/fit.test.bytrain.fucci.nobin.rds")
fit.test.insample.fucci.nobin <- cycle.npreg.insample(Y = Y_test,
theta = theta_test,
nbins = NULL, ncores=10)
saveRDS(fit.test.insample.fucci.nobin,
"../output/method-npreg.Rmd/fit.test.insample.fucci.nobin.rds")
fit.test.insample.fucci.bin <- cycle.npreg.insample(Y = Y_test,
theta = theta_test,
nbins = 100, ncores=10)
saveRDS(fit.test.insample.fucci.bin,
"../output/method-npreg.Rmd/fit.test.insample.fucci.bin.rds")
fit.train.bin <- cycle.npreg.insample(Y = Y_train,
theta = theta_train,
nbins = 100, ncores=10)
saveRDS(fit.train.bin, "../output/method-npreg.Rmd/fit.train.bin.rds")
fit.test.bytrain.fucci.bin <- cycle.npreg.outsample(Y_test,
theta_est=fit.train.bin$cell_times_est,
mu_est=fit.train.bin$mu_est,
sigma_est=fit.train.bin$sigma_est)
saveRDS(fit.test.bytrain.fucci.bin,
"../output/method-npreg.Rmd/fit.test.bytrain.fucci.bin.rds")fit.test.bytrain.fucci.nobin <- readRDS("../output/method-npreg.Rmd/fit.test.bytrain.fucci.nobin.rds")
fit.test.bytrain.fucci.bin <- readRDS("../output/method-npreg.Rmd/fit.test.bytrain.fucci.bin.rds")
fit.test.insample.fucci <- readRDS("../output/method-npreg.Rmd/fit.test.insample.fucci.rds")
fit.test.insample.fucci.bin <- readRDS("../output/method-npreg.Rmd/fit.test.insample.fucci.bin.rds")
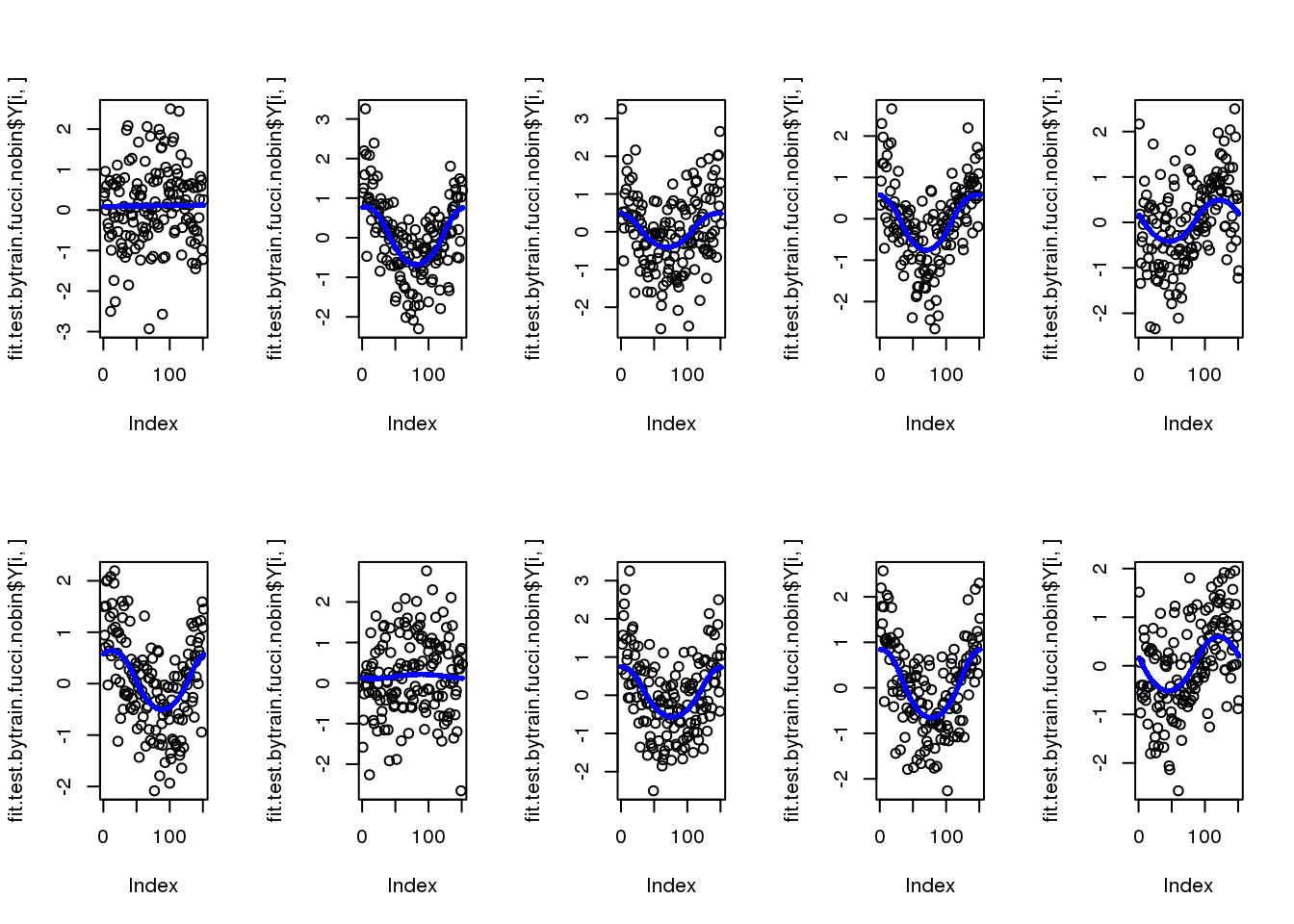
par(mfcol=c(2,5))
for (i in 1:10) {
plot(fit.test.bytrain.fucci.nobin$Y[i,])
points(fit.test.bytrain.fucci.nobin$mu_est[i,], col = "blue", cex=.6, pch=16)
}
par(mfcol=c(2,5))
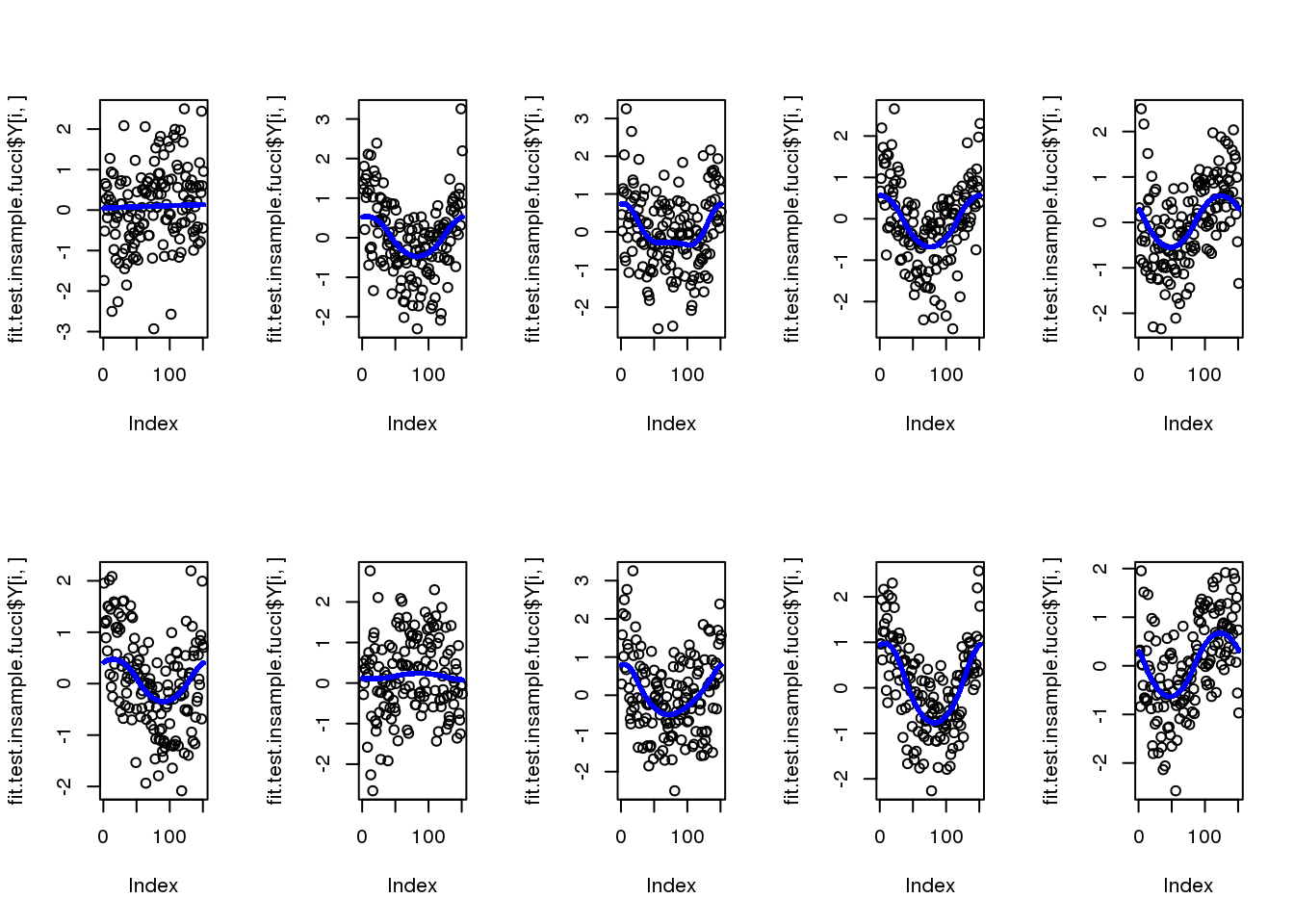
for (i in 1:10) {
plot(fit.test.insample.fucci$Y[i,])
points(fit.test.insample.fucci$mu_est[i,], col = "blue", cex=.6, pch=16)
}
fit.test.insample.fucci.bin$loglik_est[1] -1819.002fit.test.bytrain.fucci.bin$loglik_est[1] -1749.787Correlation between cell time label: use grid points to estimate gene means in the training sample
# JSTestRand(fit.test.bytrain.fucci.nobin$cell_times_est,
# theta_test[match(names(fit.test.bytrain.fucci.nobin$cell_times_est),
# names(theta_test))], NR=9999)
# JSTestRand(fit.test.insample.fucci.nobin$cell_times_est,
# theta_test[match(names(fit.test.insample.fucci.nobin$cell_times_est),
# names(theta_test))], NR=9999)Correlation between cell time labels: not use grid points to estimate gene means in the training sample
# JSTestRand(fit.test.bytrain.fucci.bin$cell_times_est,
# theta_test[match(names(fit.test.bytrain.fucci.bin$cell_times_est),
# names(theta_test))], NR=9999)
# JSTestRand(fit.test.insample.fucci.bin$cell_times_est,
# theta_test[match(names(fit.test.insample.fucci.bin$cell_times_est),
# names(theta_test))], NR=9999)Develop a classification performance measure
For every sample i in the reference label, identify the nearst k labels at a neighborhood symmetric with respect to the sample i, such that they are at [i-k, i+k]
Then, for every sample i in the set for testing, consider also the k nearest neighbors as above.
Compute for every sample i the fraction of samples in the neighborhood that are in the reference neighborhood, and the fraction of samples that are not in the neighborhood that are in the reference neighborhood. The ratio of these two gives the fold enrichment of the neighborhood samples that are in the reference neighborhood.
circ.dist.neighbors <- function(labels, k) {
mat_neighbors <- matrix(0, ncol=length(labels), nrow=length(labels))
colnames(mat_neighbors) <- labels
rownames(mat_neighbors) <- labels
N <- length(labels)
band <- round(k/2)
for (i in 1:ncol(mat_neighbors)) {
if (i == 1) {
neighbors <- c(labels[c((N-band+i):N)], labels[c((i+1):(i+band))])
mat_neighbors[,i] <- rownames(mat_neighbors) %in% neighbors
}
if (i > 1 & i <= band) {
neighbors <- c(labels[c(1:(i-1), c((N-band+i):N))], labels[c((i+1):(i+band))])
mat_neighbors[,i] <- rownames(mat_neighbors) %in% neighbors
}
if (i > band & i <= (N-band)) {
neighbors <- c(labels[c((i-band):(i-1))], labels[c((i+1):(i+band))])
mat_neighbors[,i] <- rownames(mat_neighbors) %in% neighbors
}
if (i > (N-band)) {
neighbors <- c(labels[c((i-band):(i-1))], labels[c((i+1):N, (band -(N-i)):1)])
mat_neighbors[,i] <- rownames(mat_neighbors) %in% neighbors
}
if (i == N) {
neighbors <- c(labels[c((N-band):(N-1))], labels[c(1:band)])
mat_neighbors[,i] <- rownames(mat_neighbors) %in% neighbors
}
}
return(mat_neighbors)
}
## test the code
labels_to_test <- names(theta_test)
labels_ref <- names(theta_test)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
})
names(dist_enrich) <- rownames(dist_mat_ref)
summary(dist_enrich)
## try on some data
labels_to_test <- names(fit.test.bytrain.fucci.nobin$cell_times_est)
labels_ref <- names(theta_test)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
})
names(dist_enrich) <- rownames(dist_mat_ref)
summary(dist_enrich)
# compare with predictions obtained from training sample (binned)
labels_to_test <- names(fit.test.bytrain.fucci.bin$cell_times_est)
labels_ref <- names(theta_test)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
})
names(dist_enrich) <- rownames(dist_mat_ref)
summary(dist_enrich)
# compare with predictions obtained from training sample (binned)
labels_to_test <- names(fit.test.insample.fucci.nobin$cell_times_est)
labels_ref <- names(theta_test)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
})
names(dist_enrich) <- rownames(dist_mat_ref)
summary(dist_enrich)
# compare with predictions obtained from in-sample prediction (binned)
labels_to_test <- names(fit.test.insample.fucci.bin$cell_times_est)
labels_ref <- names(theta_test)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
})
names(dist_enrich) <- rownames(dist_mat_ref)
summary(dist_enrich)Running on a larger dataset
Will run the job on cluster. See code in code/method-npreg.Rmd.
df <- readRDS(file="../data/eset-final.rds")
pdata <- pData(df)
fdata <- fData(df)
# select endogeneous genes
counts <- exprs(df)[grep("ENSG", rownames(df)), ]
log2cpm.all <- t(log2(1+(10^6)*(t(counts)/pdata$molecules)))
macosko <- readRDS("../data/cellcycle-genes-previous-studies/rds/macosko-2015.rds")
log2cpm.all <- log2cpm.all[,order(pdata$theta)]
pdata <- pdata[order(pdata$theta),]
log2cpm.quant <- readRDS("../output/npreg-trendfilter-quantile.Rmd/log2cpm.quant.rds")
# import previously identifid cell cycle genes
# cyclegenes <- readRDS("../output/npreg-methods.Rmd/cyclegenes.rds")
# cyclegenes.names <- colnames(cyclegenes)[2:6]
# select external validation samples
set.seed(99)
nvalid <- round(ncol(log2cpm.quant)*.15)
ii.valid <- sample(1:ncol(log2cpm.quant), nvalid, replace = F)
ii.nonvalid <- setdiff(1:ncol(log2cpm.quant), ii.valid)
log2cpm.quant.nonvalid <- log2cpm.quant[,ii.nonvalid]
log2cpm.quant.valid <- log2cpm.quant[,ii.valid]
theta <- pdata$theta
names(theta) <- rownames(pdata)
theta.nonvalid <- theta[ii.nonvalid]
theta.valid <- theta[ii.valid]
sig.genes <- readRDS("../output/npreg-trendfilter-quantile.Rmd/out.stats.ordered.sig.rds")
expr.sig <- log2cpm.quant.nonvalid[rownames(log2cpm.quant.nonvalid) %in% rownames(sig.genes)[1:10], ]
# set training samples
source("../peco/R/primes.R")
source("../peco/R/partitionSamples.R")
parts <- partitionSamples(1:ncol(log2cpm.quant.nonvalid), runs=5,
nsize.each = rep(151,5))
part_indices <- parts$partitionsfold.train <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.train.fold.",fold,".rds")) })
fold.test.bytrain.fucci <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.test.bytrain.fucci.fold.", fold, ".rds"))})
fold.test.insample.fucci <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.test.insample.fucci.fold.",fold,".rds"))})
do.call(rbind, lapply(1:5, function(f) {
data.frame(train.fucci=fold.train[[f]]$loglik_est/4,
test.bytrain.fucci=fold.test.bytrain.fucci[[f]]$loglik_est,
test.insample.fucci=fold.test.insample.fucci[[f]]$loglik_est)
})) train.fucci test.bytrain.fucci test.insample.fucci
1 -20409.85 -19779.37 -20199.67
2 -20320.87 -19981.03 -20236.37
3 -20304.71 -20033.21 -20459.61
4 -20380.62 -20011.64 -20362.36
5 -20257.38 -20077.28 -20775.22compute two measures to evaluate the results:
correlation between labels
fold enrichment of neighborhood samples defined in the reference label
source("../code/utility.R")
eval_cor <- vector("list", 5)
eval_neighbor <- vector("list", 5)
for (f in 1:5) {
eval_cor[[f]] <- JSTestRand(theta.nonvalid[part_indices[[f]]$test],
fold.test.bytrain.fucci[[f]]$cell_times_est, NR=9999)
}
eval_cor <- do.call(rbind, eval_cor)
colnames(eval_cor) <- c("rho", "pval")
for (f in 1:5) {
labels_ref <- names(theta.nonvalid[part_indices[[f]]$test])
labels_to_test <- names(fold.test.bytrain.fucci[[f]]$cell_times_est)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
N <- length(labels_ref)
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
# labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
#prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
#prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
# return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
TP <- sum(dist_mat_ref[i,]==1 & dist_mat_to_test[i,]==1)
FP <- sum(dist_mat_ref[i,]==0 & dist_mat_to_test[i,]==1)
FN <- sum(dist_mat_ref[i,]==1 & dist_mat_to_test[i,]==0)
precision <- TP/(TP+FP)
recall <- TP/(TP+FN)
if ((precision+recall)==0) {
F1score <- 0
} else {
F1score <- 2*precision*recall/(precision+recall)
}
return(F1score)
})
names(dist_enrich) <- rownames(dist_mat_ref)
eval_neighbor[[f]] <- dist_enrich
}
saveRDS(eval_cor, "../output/method-npreg.Rmd/eval_cor.rds")
saveRDS(eval_neighbor, "../output/method-npreg.Rmd/eval_neighbor.rds")eval_cor <- readRDS("../output/method-npreg.Rmd/eval_cor.rds")
eval_neighbor <- readRDS("../output/method-npreg.Rmd/eval_neighbor.rds")
eval_cor rho pval
[1,] 0.6143964 0.0001
[2,] -0.2054608 0.0111
[3,] -0.3310730 0.0001
[4,] -0.4243352 0.0001
[5,] -0.2633038 0.0008lapply(eval_neighbor, function(x) table(x))[[1]]
x
0 0.1 0.2 0.3 0.4 0.5
59 47 27 14 3 1
[[2]]
x
0 0.1 0.2 0.3 0.4
64 37 38 11 1
[[3]]
x
0 0.1 0.2 0.3 0.4
62 49 34 5 1
[[4]]
x
0 0.1 0.2 0.3 0.4
53 55 30 11 2
[[5]]
x
0 0.1 0.2 0.3 0.4
44 57 29 17 4 eval_neighbor_table <- matrix(0, nrow=5, ncol=8)
for (i in 1:5 ) {
eval_neighbor_table[i,1:length(table(eval_neighbor[[i]]))] <- as.numeric(table(eval_neighbor[[i]]))
}
colnames(eval_neighbor_table) <- c("0", "0.1", "0.2", "0.3", "0.4", "0.5", "mean", "sd")
#eval_neighbor_table <- do.call(rbind, lapply(eval_neighbor, function(x) table(x)))
eval_neighbor_table[,7] <- sapply(eval_neighbor, function(x) round(mean(x), digits=2))
eval_neighbor_table[,8] <- sapply(eval_neighbor, function(x) round(sd(x), digits=2))
eval_neighbor_table 0 0.1 0.2 0.3 0.4 0.5 mean sd
[1,] 59 47 27 14 3 1 0.11 0.11
[2,] 64 37 38 11 1 0 0.10 0.10
[3,] 62 49 34 5 1 0 0.09 0.09
[4,] 53 55 30 11 2 0 0.10 0.10
[5,] 44 57 29 17 4 0 0.12 0.11Running on 476 genes
Will run the job on cluster. See code in code/method-npreg.Rmd.
df <- readRDS(file="../data/eset-final.rds")
pdata <- pData(df)
fdata <- fData(df)
# select endogeneous genes
counts <- exprs(df)[grep("ENSG", rownames(df)), ]
log2cpm.all <- t(log2(1+(10^6)*(t(counts)/pdata$molecules)))
macosko <- readRDS("../data/cellcycle-genes-previous-studies/rds/macosko-2015.rds")
log2cpm.all <- log2cpm.all[,order(pdata$theta)]
pdata <- pdata[order(pdata$theta),]
log2cpm.quant <- readRDS("../output/npreg-trendfilter-quantile.Rmd/log2cpm.quant.rds")
# import previously identifid cell cycle genes
# cyclegenes <- readRDS("../output/npreg-methods.Rmd/cyclegenes.rds")
# cyclegenes.names <- colnames(cyclegenes)[2:6]
# select external validation samples
set.seed(99)
nvalid <- round(ncol(log2cpm.quant)*.15)
ii.valid <- sample(1:ncol(log2cpm.quant), nvalid, replace = F)
ii.nonvalid <- setdiff(1:ncol(log2cpm.quant), ii.valid)
log2cpm.quant.nonvalid <- log2cpm.quant[,ii.nonvalid]
log2cpm.quant.valid <- log2cpm.quant[,ii.valid]
theta <- pdata$theta
names(theta) <- rownames(pdata)
theta.nonvalid <- theta[ii.nonvalid]
theta.valid <- theta[ii.valid]
# sig.genes <- readRDS("../output/npreg-trendfilter-quantile.Rmd/out.stats.ordered.sig.rds")
# expr.sig <- log2cpm.quant.nonvalid[rownames(log2cpm.quant.nonvalid) %in% rownames(sig.genes)[1:10], ]
# set training samples
source("../peco/R/primes.R")
source("../peco/R/partitionSamples.R")
parts <- partitionSamples(1:ncol(log2cpm.quant.nonvalid), runs=5,
nsize.each = rep(151,5))
part_indices <- parts$partitionsfold.train <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.train.fold.",fold,".all.rds")) })
fold.test.bytrain.fucci <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.test.bytrain.fucci.fold.", fold, ".all.rds"))})
fold.test.insample.fucci <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.test.insample.fucci.fold.",fold,".all.rds"))})
do.call(rbind, lapply(1:5, function(f) {
data.frame(train.fucci=fold.train[[f]]$loglik_est/4,
test.bytrain.fucci=fold.test.bytrain.fucci[[f]]$loglik_est,
test.insample.fucci=fold.test.insample.fucci[[f]]$loglik_est)
})) train.fucci test.bytrain.fucci test.insample.fucci
1 -100377.87 -98241.85 -99559.33
2 -100484.07 -98575.41 -99255.19
3 -99993.06 -100156.22 -100518.49
4 -100207.10 -99117.05 -100321.46
5 -100016.10 -100284.54 -101743.31compute two measures to evaluate the results:
correlation between labels
fold enrichment of neighborhood samples defined in the reference label
source("../code/utility.R")
eval_cor <- vector("list", 5)
eval_neighbor <- vector("list", 5)
for (f in 1:5) {
eval_cor[[f]] <- JSTestRand(theta.nonvalid[part_indices[[f]]$test],
fold.test.bytrain.fucci[[f]]$cell_times_est, NR=9999)
}
eval_cor <- do.call(rbind, eval_cor)
colnames(eval_cor) <- c("rho", "pval")
for (f in 1:5) {
labels_ref <- names(theta.nonvalid[part_indices[[f]]$test])
labels_to_test <- names(fold.test.bytrain.fucci[[f]]$cell_times_est)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
N <- length(labels_ref)
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
# labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
#prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
#prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
# return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
TP <- sum(dist_mat_ref[i,]==1 & dist_mat_to_test[i,]==1)
FP <- sum(dist_mat_ref[i,]==0 & dist_mat_to_test[i,]==1)
FN <- sum(dist_mat_ref[i,]==1 & dist_mat_to_test[i,]==0)
precision <- TP/(TP+FP)
recall <- TP/(TP+FN)
if ((precision+recall)==0) {
F1score <- 0
} else {
F1score <- 2*precision*recall/(precision+recall)
}
return(F1score)
})
names(dist_enrich) <- rownames(dist_mat_ref)
eval_neighbor[[f]] <- dist_enrich
}
saveRDS(eval_cor, "../output/method-npreg.Rmd/eval_cor.all.rds")
saveRDS(eval_neighbor, "../output/method-npreg.Rmd/eval_neighbor.all.rds")eval_cor <- readRDS("../output/method-npreg.Rmd/eval_cor.all.rds")
eval_neighbor <- readRDS("../output/method-npreg.Rmd/eval_neighbor.all.rds")
eval_cor rho pval
[1,] 0.6289235 1e-04
[2,] -0.2695323 8e-04
[3,] -0.3471673 2e-04
[4,] -0.3806333 1e-04
[5,] -0.2256917 6e-03sapply(eval_neighbor, function(x) table(x))[[1]]
x
0 0.1 0.2 0.3 0.4
65 50 27 6 3
[[2]]
x
0 0.1 0.2 0.3 0.4
69 56 18 6 2
[[3]]
x
0 0.1 0.2 0.3
58 53 31 9
[[4]]
x
0 0.1 0.2 0.3 0.4
57 46 37 10 1
[[5]]
x
0 0.1 0.2 0.3 0.4
51 60 31 8 1 eval_neighbor_table <- matrix(0, nrow=5, ncol=7)
for (i in 1:5 ) {
eval_neighbor_table[i,1:length(table(eval_neighbor[[i]]))] <- as.numeric(table(eval_neighbor[[i]]))
}
colnames(eval_neighbor_table) <- c("0", "0.1", "0.2", "0.3", "0.4", "mean", "sd")
#eval_neighbor_table <- do.call(rbind, lapply(eval_neighbor, function(x) table(x)))
eval_neighbor_table[,6] <- sapply(eval_neighbor, function(x) round(mean(x), digits=2))
eval_neighbor_table[,7] <- sapply(eval_neighbor, function(x) round(sd(x), digits=2))
eval_neighbor_table 0 0.1 0.2 0.3 0.4 mean sd
[1,] 65 50 27 6 3 0.09 0.10
[2,] 69 56 18 6 2 0.08 0.09
[3,] 58 53 31 9 0 0.09 0.09
[4,] 57 46 37 10 1 0.10 0.10
[5,] 51 60 31 8 1 0.10 0.09Running on Macosko genes
Will run the job on cluster. See code in code/method-npreg.Rmd.
df <- readRDS(file="../data/eset-final.rds")
pdata <- pData(df)
fdata <- fData(df)
# select endogeneous genes
counts <- exprs(df)[grep("ENSG", rownames(df)), ]
log2cpm.all <- t(log2(1+(10^6)*(t(counts)/pdata$molecules)))
macosko <- readRDS("../data/cellcycle-genes-previous-studies/rds/macosko-2015.rds")
log2cpm.all <- log2cpm.all[,order(pdata$theta)]
pdata <- pdata[order(pdata$theta),]
log2cpm.quant <- readRDS("../output/npreg-trendfilter-quantile.Rmd/log2cpm.quant.rds")
# import previously identifid cell cycle genes
# cyclegenes <- readRDS("../output/npreg-methods.Rmd/cyclegenes.rds")
# cyclegenes.names <- colnames(cyclegenes)[2:6]
# select external validation samples
set.seed(99)
nvalid <- round(ncol(log2cpm.quant)*.15)
ii.valid <- sample(1:ncol(log2cpm.quant), nvalid, replace = F)
ii.nonvalid <- setdiff(1:ncol(log2cpm.quant), ii.valid)
log2cpm.quant.nonvalid <- log2cpm.quant[,ii.nonvalid]
log2cpm.quant.valid <- log2cpm.quant[,ii.valid]
theta <- pdata$theta
names(theta) <- rownames(pdata)
theta.nonvalid <- theta[ii.nonvalid]
theta.valid <- theta[ii.valid]
# sig.genes <- readRDS("../output/npreg-trendfilter-quantile.Rmd/out.stats.ordered.sig.rds")
# expr.sig <- log2cpm.quant.nonvalid[rownames(log2cpm.quant.nonvalid) %in% rownames(sig.genes)[1:10], ]
# set training samples
source("../peco/R/primes.R")
source("../peco/R/partitionSamples.R")
parts <- partitionSamples(1:ncol(log2cpm.quant.nonvalid), runs=5,
nsize.each = rep(151,5))
part_indices <- parts$partitionsfold.train <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.train.fold.",fold,".macosko.rds")) })
fold.test.bytrain.fucci <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.test.bytrain.fucci.fold.", fold, ".macosko.rds"))})
fold.test.insample.fucci <- lapply(1:5, function(fold) {
readRDS(paste0("../output/method-npreg.Rmd/fold.test.insample.fucci.fold.",fold,".macosko.rds"))})
do.call(rbind, lapply(1:5, function(f) {
data.frame(train.fucci=fold.train[[f]]$loglik_est/4,
test.bytrain.fucci=fold.test.bytrain.fucci[[f]]$loglik_est,
test.insample.fucci=fold.test.insample.fucci[[f]]$loglik_est)
})) train.fucci test.bytrain.fucci test.insample.fucci
1 -110269.8 -109393.6 -109433.6
2 -110321.7 -109331.5 -109517.5
3 -110183.5 -110036.1 -110050.9
4 -110178.0 -109770.8 -109750.4
5 -110116.6 -110038.4 -110172.7compute two measures to evaluate the results:
correlation between labels
fold enrichment of neighborhood samples defined in the reference label
source("../code/utility.R")
eval_cor <- vector("list", 5)
eval_neighbor <- vector("list", 5)
for (f in 1:5) {
eval_cor[[f]] <- JSTestRand(theta.nonvalid[part_indices[[f]]$test],
fold.test.bytrain.fucci[[f]]$cell_times_est, NR=9999)
}
eval_cor <- do.call(rbind, eval_cor)
colnames(eval_cor) <- c("rho", "pval")
for (f in 1:5) {
labels_ref <- names(theta.nonvalid[part_indices[[f]]$test])
labels_to_test <- names(fold.test.bytrain.fucci[[f]]$cell_times_est)
dist_mat_ref <- circ.dist.neighbors(labels_ref, k=10)
dist_mat_to_test <- circ.dist.neighbors(labels_to_test, k=10)
ii.match <- match(colnames(dist_mat_ref), colnames(dist_mat_to_test))
dist_mat_to_test <- dist_mat_to_test[ii.match, ii.match]
N <- length(labels_ref)
dist_enrich <- sapply(1:N, function(i) {
lab_self <- rownames(dist_mat_ref)[i]
labs_ref <- rownames(dist_mat_ref)[which(dist_mat_ref[i,]==1)]
labs_to_test <- rownames(dist_mat_to_test)[which(dist_mat_to_test[i,]==1)]
# labs_to_test_nonneighbors <- setdiff(rownames(dist_mat_to_test), c(labs_to_test, lab_self))
#prop_neighbors_in_ref <- sum(labs_to_test %in% labs_ref)/length(labs_to_test)
#prop_nonneighbors_in_ref <- sum(labs_to_test_nonneighbors %in% labs_ref)/length(labs_to_test_nonneighbors)
# return(prop_neighbors_in_ref/prop_nonneighbors_in_ref)
TP <- sum(dist_mat_ref[i,]==1 & dist_mat_to_test[i,]==1)
FP <- sum(dist_mat_ref[i,]==0 & dist_mat_to_test[i,]==1)
FN <- sum(dist_mat_ref[i,]==1 & dist_mat_to_test[i,]==0)
precision <- TP/(TP+FP)
recall <- TP/(TP+FN)
if ((precision+recall)==0) {
F1score <- 0
} else {
F1score <- 2*precision*recall/(precision+recall)
}
return(F1score)
})
names(dist_enrich) <- rownames(dist_mat_ref)
eval_neighbor[[f]] <- dist_enrich
}
saveRDS(eval_cor, "../output/method-npreg.Rmd/eval_cor.macosko.rds")
saveRDS(eval_neighbor, "../output/method-npreg.Rmd/eval_neighbor.macosko.rds")eval_cor <- readRDS("../output/method-npreg.Rmd/eval_cor.macosko.rds")
eval_neighbor <- readRDS("../output/method-npreg.Rmd/eval_neighbor.macosko.rds")
eval_cor rho pval
[1,] 0.5968372 0.0001
[2,] -0.1700843 0.0385
[3,] -0.3336919 0.0002
[4,] -0.3536661 0.0001
[5,] -0.2281700 0.0057lapply(eval_neighbor, function(x) summary(x))[[1]]
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00000 0.00000 0.05033 0.10000 0.30000
[[2]]
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00000 0.10000 0.07152 0.10000 0.30000
[[3]]
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00000 0.10000 0.07815 0.10000 0.40000
[[4]]
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00000 0.10000 0.08079 0.10000 0.40000
[[5]]
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.00000 0.00000 0.10000 0.07682 0.10000 0.30000 lapply(eval_neighbor, function(x) table(x))[[1]]
x
0 0.1 0.2 0.3
90 48 11 2
[[2]]
x
0 0.1 0.2 0.3
67 63 18 3
[[3]]
x
0 0.1 0.2 0.3 0.4
70 55 16 9 1
[[4]]
x
0 0.1 0.2 0.3 0.4
62 64 18 6 1
[[5]]
x
0 0.1 0.2 0.3
67 54 28 2 eval_neighbor_table <- matrix(0, nrow=5, ncol=7)
for (i in 1:5 ) {
eval_neighbor_table[i,1:length(table(eval_neighbor[[i]]))] <- as.numeric(table(eval_neighbor[[i]]))
}
colnames(eval_neighbor_table) <- c("0", "0.1", "0.2", "0.3", "0.4", "mean", "sd")
#eval_neighbor_table <- do.call(rbind, lapply(eval_neighbor, function(x) table(x)))
eval_neighbor_table[,6] <- sapply(eval_neighbor, function(x) round(mean(x),digits=2))
eval_neighbor_table[,7] <- sapply(eval_neighbor, function(x) round(sd(x),digits=2))
eval_neighbor_table 0 0.1 0.2 0.3 0.4 mean sd
[1,] 90 48 11 2 0 0.05 0.07
[2,] 67 63 18 3 0 0.07 0.08
[3,] 70 55 16 9 1 0.08 0.09
[4,] 62 64 18 6 1 0.08 0.08
[5,] 67 54 28 2 0 0.08 0.08Session information
sessionInfo()R version 3.4.3 (2017-11-30)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Scientific Linux 7.4 (Nitrogen)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.2.19-el7-x86_64/lib/libopenblas_haswellp-r0.2.19.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] Biobase_2.38.0 BiocGenerics_0.24.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.17 digest_0.6.15 rprojroot_1.3-2 backports_1.1.2
[5] git2r_0.21.0 magrittr_1.5 evaluate_0.10.1 stringi_1.1.6
[9] rmarkdown_1.10 tools_3.4.3 stringr_1.2.0 yaml_2.1.16
[13] compiler_3.4.3 htmltools_0.3.6 knitr_1.20 This R Markdown site was created with workflowr