Overlap molQTLs, Opposite Direction
Briana Mittleman
10/8/2018
Last updated: 2018-10-09
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 605aa2d
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: output/.DS_Store Untracked files: Untracked: KalistoAbundance18486.txt Untracked: analysis/genometrack_figs.Rmd Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed Untracked: analysis/snake.config.notes.Rmd Untracked: analysis/verifyBAM.Rmd Untracked: data/18486.genecov.txt Untracked: data/APApeaksYL.total.inbrain.bed Untracked: data/NuclearApaQTLs.txt Untracked: data/RNAkalisto/ Untracked: data/TotalApaQTLs.txt Untracked: data/Totalpeaks_filtered_clean.bed Untracked: data/YL-SP-18486-T-combined-genecov.txt Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt Untracked: data/apaExamp/ Untracked: data/bedgraph_peaks/ Untracked: data/bin200.5.T.nuccov.bed Untracked: data/bin200.Anuccov.bed Untracked: data/bin200.nuccov.bed Untracked: data/clean_peaks/ Untracked: data/comb_map_stats.csv Untracked: data/comb_map_stats.xlsx Untracked: data/comb_map_stats_39ind.csv Untracked: data/combined_reads_mapped_three_prime_seq.csv Untracked: data/ensemble_to_genename.txt Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed Untracked: data/first50lines_closest.txt Untracked: data/gencov.test.csv Untracked: data/gencov.test.txt Untracked: data/gencov_zero.test.csv Untracked: data/gencov_zero.test.txt Untracked: data/gene_cov/ Untracked: data/joined Untracked: data/leafcutter/ Untracked: data/merged_combined_YL-SP-threeprimeseq.bg Untracked: data/mol_overlap/ Untracked: data/nom_QTL/ Untracked: data/nom_QTL_opp/ Untracked: data/nom_QTL_trans/ Untracked: data/nuc6up/ Untracked: data/other_qtls/ Untracked: data/peakPerRefSeqGene/ Untracked: data/perm_QTL/ Untracked: data/perm_QTL_opp/ Untracked: data/perm_QTL_trans/ Untracked: data/reads_mapped_three_prime_seq.csv Untracked: data/smash.cov.results.bed Untracked: data/smash.cov.results.csv Untracked: data/smash.cov.results.txt Untracked: data/smash_testregion/ Untracked: data/ssFC200.cov.bed Untracked: data/temp.file1 Untracked: data/temp.file2 Untracked: data/temp.gencov.test.txt Untracked: data/temp.gencov_zero.test.txt Untracked: output/picard/ Untracked: output/plots/ Untracked: output/qual.fig2.pdf Unstaged changes: Modified: analysis/28ind.peak.explore.Rmd Modified: analysis/39indQC.Rmd Modified: analysis/PeakToGeneAssignment.Rmd Modified: analysis/cleanupdtseq.internalpriming.Rmd Modified: analysis/dif.iso.usage.leafcutter.Rmd Modified: analysis/diff_iso_pipeline.Rmd Modified: analysis/explore.filters.Rmd Modified: analysis/overlapMolQTL.Rmd Modified: analysis/overlap_qtls.Rmd Modified: analysis/peakOverlap_oppstrand.Rmd Modified: analysis/pheno.leaf.comb.Rmd Modified: analysis/swarmPlots_QTLs.Rmd Modified: analysis/test.max2.Rmd Modified: code/Snakefile
Expand here to see past versions:
In the OverlapMolQTL analysis I looked at significant molecular QTLs and asked if they are also significant snp:gene pairs in the ApaQTLs. In this analysis, I will look at the significant ApaQTLs and ask if the snp:gene pairs are significant in the other molecular phenotypes. I expect enrichment of low pvalues in protQTLs but less in RNA.
I am going to complete this analysis first for the totalAPA QTLs.
library(workflowr)This is workflowr version 1.1.1
Run ?workflowr for help getting startedlibrary(reshape2)
library(tidyverse)── Attaching packages ─────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.0.0 ✔ purrr 0.2.5
✔ tibble 1.4.2 ✔ dplyr 0.7.6
✔ tidyr 0.8.1 ✔ stringr 1.3.1
✔ readr 1.1.1 ✔ forcats 0.3.0── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(VennDiagram)Loading required package: gridLoading required package: futile.loggerlibrary(data.table)
Attaching package: 'data.table'The following objects are masked from 'package:dplyr':
between, first, lastThe following object is masked from 'package:purrr':
transposeThe following objects are masked from 'package:reshape2':
dcast, meltlibrary(qvalue)
set.seed(327)MolQTL pvalues for Total ApaQTLs
sigTotAPAinMolPheno.R
#!/bin/rscripts
#this script creates takes in the permuted APAQTL results for the total fraction and nominal pvalues from the molecular phenotpye molecular phenotype
library(dplyr)
library(tidyr)
library(ggplot2)
library(readr)
library(optparse)
geneNames=read.table("/project2/gilad/briana/genome_anotation_data/ensemble_to_genename.txt", sep="\t", header=T, stringsAsFactors = F)
tot_perm=read.table("/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_transcript_permResBH.txt", header = T,stringsAsFactors=F)
sigSNPgene=tot_perm %>% filter(-log10(bh)>1) %>% separate(pid, into=c("chr", "start", "end", "id"), sep=":") %>% separate(id, into=c("Gene.name", "strand", "peaknum"), sep="_") %>% dplyr::select(Gene.name, sid, bh) %>% group_by(Gene.name) %>% top_n(-1, bh) %>% ungroup() %>% dplyr::select(Gene.name, sid)
option_list = list(
make_option(c("-M", "--molNom"), action="store", default=NA, type='character', help="molecular Nom results"),
make_option(c("-O", "--output"), action="store", default=NA, type='character', help="output file for total APA sig snps in mol qtl")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
if (opt$molNom == "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_prot.fixed.nominal.out") {
in_file=read.table(opt$molNom, col.names = c("Gene.stable.ID", "sid", "dist", "pval", "slope"),stringsAsFactors=F)
file_newNames=in_file %>% inner_join(geneNames, by="Gene.stable.ID") %>% dplyr::select("Gene.name", "sid", "pval")
} else {
in_file=read.table(opt$molNom, col.names = c("pid", "sid", "dist", "pval", "slope"),stringsAsFactors=F)
file_newNames=in_file %>% separate(pid, into=c("Gene.stable.ID", "ver"), sep ="[.]") %>% inner_join(geneNames, by="Gene.stable.ID") %>% dplyr::select("Gene.name", "sid", "pval")
}
overlap= file_newNames %>% semi_join(sigSNPgene, by=c("Gene.name", "sid"))
write.table(overlap, file=opt$output, quote=F, col.names = T, row.names = F)Run this first on the rnaQTLs.
run_sigTotAPAinMolPhenoRNA.sh
#!/bin/bash
#SBATCH --job-name=run_sigTotAPAinMolPhenoRNA
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigTotAPAinMolPhenoRNA.out
#SBATCH --error=run_sigTotAPAinMolPhenoRNA.err
#SBATCH --partition=bigmem2
#SBATCH --mem=64G
#SBATCH --mail-type=END
module load R
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPvalRNA.txt" run_sigTotAPAinMolPhenoProt.sh
#!/bin/bash
#SBATCH --job-name=run_sigTotAPAinMolPhenoProt
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigTotAPAinMolPhenoProt.out
#SBATCH --error=run_sigTotAPAinMolPhenoProt.err
#SBATCH --partition=bigmem2
#SBATCH --mem=64G
#SBATCH --mail-type=END
module load R
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_prot.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPvalProtein.txt"
run_sigTotAPAinMolPhenoProt.sh
#!/bin/bash
#SBATCH --job-name=run_sigTotAPAinMolPhenoProt
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigTotAPAinMolPhenoProt.out
#SBATCH --error=run_sigTotAPAinMolPhenoProt.err
#SBATCH --partition=bigmem2
#SBATCH --mem=64G
#SBATCH --mail-type=END
module load R
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_prot.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPvalProtein.txt"
run_sigTotAPAinMolPhenoRNAg.sh
#!/bin/bash
#SBATCH --job-name=run_sigTotAPAinMolPhenoRNAg
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigTotAPAinMolPhenoRNAg.out
#SBATCH --error=run_sigTotAPAinMolPhenoRNAg.err
#SBATCH --partition=bigmem2
#SBATCH --mem=64G
#SBATCH --mail-type=END
module load R
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_RNAseqGeuvadis.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPvalRNAg.txt"
run_sigTotAPAinMolPhenoRibo.sh
#!/bin/bash
#SBATCH --job-name=run_sigTotAPAinMolPhenoRibo
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigTotAPAinMolPhenoRibo.out
#SBATCH --error=run_sigTotAPAinMolPhenoRibo.err
#SBATCH --partition=bigmem2
#SBATCH --mem=64G
#SBATCH --mail-type=END
module load R
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_ribo_phase2.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPvalribo.txt"
run_sigTotAPAinMolPheno4su.sh
#!/bin/bash
#SBATCH --job-name=run_sigTotAPAinMolPheno4su
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigTotAPAinMolPheno4su.out
#SBATCH --error=run_sigTotAPAinMolPheno4su.err
#SBATCH --partition=bigmem2
#SBATCH --mem=64G
#SBATCH --mail-type=END
module load R
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_4su30.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPval4su30.txt"
Rscript sigTotAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_4su60.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molTotal/TotAPAqtlsPval4su60.txt" MolQTL pvalues for Nuclear ApaQTLs
sigNucAPAinMolPheno.R
#!/bin/rscripts
#this script creates takes in the permuted APAQTL results for the total fraction and nominal pvalues from the molecular phenotpye molecular phenotype
library(dplyr)
library(tidyr)
library(ggplot2)
library(readr)
library(optparse)
geneNames=read.table("/project2/gilad/briana/genome_anotation_data/ensemble_to_genename.txt", sep="\t", header=T, stringsAsFactors = F)
nuc_perm=read.table("/project2/gilad/briana/threeprimeseq/data/perm_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_transcript_permResBH.txt", header = T,stringsAsFactors=F)
sigSNPgene=nuc_perm %>% filter(-log10(bh)>1) %>% separate(pid, into=c("chr", "start", "end", "id"), sep=":") %>% separate(id, into=c("Gene.name", "strand", "peaknum"), sep="_") %>% dplyr::select(Gene.name, sid, bh) %>% group_by(Gene.name) %>% top_n(-1, bh) %>% ungroup() %>% dplyr::select(Gene.name, sid)
option_list = list(
make_option(c("-M", "--molNom"), action="store", default=NA, type='character', help="molecular Nom results"),
make_option(c("-O", "--output"), action="store", default=NA, type='character', help="output file for total APA sig snps in mol qtl")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
if (opt$molNom == "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_prot.fixed.nominal.out") {
in_file=read.table(opt$molNom, col.names = c("Gene.stable.ID", "sid", "dist", "pval", "slope"),stringsAsFactors=F)
file_newNames=in_file %>% inner_join(geneNames, by="Gene.stable.ID") %>% dplyr::select("Gene.name", "sid", "pval")
} else {
in_file=read.table(opt$molNom, col.names = c("pid", "sid", "dist", "pval", "slope"),stringsAsFactors=F)
file_newNames=in_file %>% separate(pid, into=c("Gene.stable.ID", "ver"), sep ="[.]") %>% inner_join(geneNames, by="Gene.stable.ID") %>% dplyr::select("Gene.name", "sid", "pval")
}
overlap= file_newNames %>% semi_join(sigSNPgene, by=c("Gene.name", "sid"))
write.table(overlap, file=opt$output, quote=F, col.names = T, row.names = F)1 bash script for all of the phenotypes
run_sigNucAPAinMolPheno.sh
#!/bin/bash
#SBATCH --job-name=run_sigNucAPAinMolPheno
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_sigNucAPAinMolPheno.out
#SBATCH --error=run_sigNucAPAinMolPheno.err
#SBATCH --partition=broadwl
#SBATCH --mem=32G
#SBATCH --mail-type=END
module load R
Rscript sigNucAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_RNAseq_phase2.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molNuclear/NucAPAqtlsPvalRNA.txt"
Rscript sigNucAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_prot.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molNuclear/NucAPAqtlsPvalProtein.txt"
Rscript sigNucAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_RNAseqGeuvadis.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molNuclear/NucAPAqtlsPvalRNAg.txt"
Rscript sigNucAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_ribo_phase2.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molNuclear/NucAPAqtlsPvalribo.txt"
Rscript sigNucAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_4su30.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molNuclear/NucAPAqtlsPval4su30.txt"
Rscript sigNucAPAinMolPheno.R --molNom "/project2/gilad/briana/threeprimeseq/data/molecular_QTLs/nom/fastqtl_qqnorm_4su60.fixed.nominal.out" --output "/project2/gilad/briana/threeprimeseq/data/molecular_overlap/APA2molNuclear/NucAPAqtlsPval4su60.txt"
Create Histograms
Total
I will next estimate sharing with pi_1 and create histograms of the resulting pvalues.
- Protein
totAPAinProt=read.table("../data/mol_overlap/APA2molTotal/TotAPAqtlsPvalProtein.txt", header = T, stringsAsFactors = F)
qval_prot=pi0est(totAPAinProt$pval, pi0.method = "bootstrap")- RNA
totAPAinRNA=read.table("../data/mol_overlap/APA2molTotal/TotAPAqtlsPvalRNA.txt", header = T, stringsAsFactors = F)
qval_RNA=pi0est(totAPAinRNA$pval, pi0.method = "bootstrap")- RNA Geuvadis
totAPAinRNAg=read.table("../data/mol_overlap/APA2molTotal/TotAPAqtlsPvalRNAg.txt", header = T, stringsAsFactors = F)
qval_RNAg=pi0est(totAPAinRNAg$pval, pi0.method = "bootstrap")*Ribo
totAPAinRibo=read.table("../data/mol_overlap/APA2molTotal/TotAPAqtlsPvalribo.txt", header = T, stringsAsFactors = F)
qval_Ribo=pi0est(totAPAinRibo$pval, pi0.method = "bootstrap")- 4su30
totAPAinsu30=read.table("../data/mol_overlap/APA2molTotal/TotAPAqtlsPval4su30.txt", header = T, stringsAsFactors = F)
qval_su30=pi0est(totAPAinsu30$pval, pi0.method = "bootstrap")- 4su60
totAPAinsu60=read.table("../data/mol_overlap/APA2molTotal/TotAPAqtlsPval4su60.txt", header = T, stringsAsFactors = F)
qval_su60=pi0est(totAPAinsu60$pval, pi0.method = "bootstrap")All plots:
par(mfrow=c(2,3))
hist(totAPAinsu30$pval, xlab="4su30 Pvalue", main="Significant Total APA QTLs \n 4su30")
text(.6,15, paste("pi_1=", round((1-qval_su30$pi0), digit=3), sep=" "))
hist(totAPAinsu60$pval, xlab="4su60 Pvalue", main="Significant Total APA QTLs \n 4su60")
text(.6,15, paste("pi_1=", round((1-qval_su60$pi0), digit=3), sep=" "))
hist(totAPAinRNA$pval, xlab="RNAPvalue", main="Significant Total APA QTLs \n RNA")
text(.6,18, paste("pi_1=", round((1-qval_RNA$pi0), digit=3), sep=" "))
hist(totAPAinRNAg$pval, xlab="RNA Guevadis Pvalue", main="Significant Total APA QTLs \n RNA Geuvadis")
text(.6,18, paste("pi_1=", round((1-qval_RNAg$pi0), digit=3), sep=" "))
hist(totAPAinRibo$pval, xlab="Ribo (Translation) Pvalue", main="Significant Total APA QTLs \n Ribo")
text(.6,15, paste("pi_1=", round((1-qval_Ribo$pi0), digit=3), sep=" "))
hist(totAPAinProt$pval, xlab="Protein Pvalue", main="Significant Total APA QTLs \n Protein")
text(.6,10, paste("pi_1=", round((1-qval_prot$pi0), digit=3), sep=" "))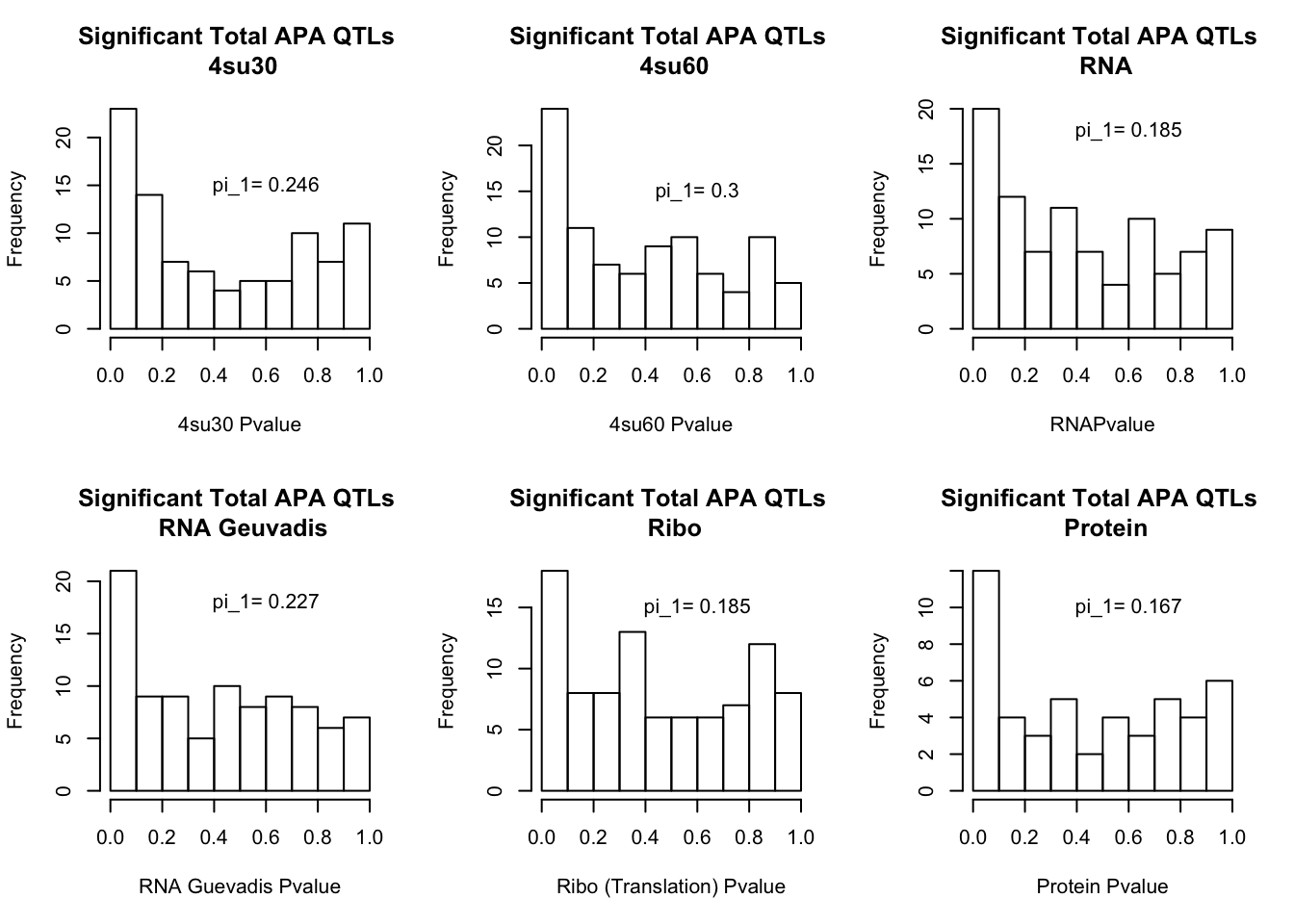
Nuclear
I will next estimate sharing with pi_1 and create histograms of the resulting pvalues.
- Protein
NucAPAinProt=read.table("../data/mol_overlap/APA2molNuclear/NucAPAqtlsPvalProtein.txt", header = T, stringsAsFactors = F)
qval_protN=pi0est(NucAPAinProt$pval, pi0.method = "bootstrap")- RNA
NucAPAinRNA=read.table("../data/mol_overlap/APA2molNuclear/NucAPAqtlsPvalRNA.txt", header = T, stringsAsFactors = F)
qval_RNAN=pi0est(NucAPAinRNA$pval, pi0.method = "bootstrap")- RNA Geuvadis
NucAPAinRNAg=read.table("../data/mol_overlap/APA2molNuclear/NucAPAqtlsPvalRNAg.txt", header = T, stringsAsFactors = F)
qval_RNAgN=pi0est(NucAPAinRNAg$pval, pi0.method = "bootstrap")*Ribo
NucAPAinRibo=read.table("../data/mol_overlap/APA2molNuclear/NucAPAqtlsPvalribo.txt", header = T, stringsAsFactors = F)
qval_RiboN=pi0est(NucAPAinRibo$pval, pi0.method = "bootstrap")- 4su30
NucAPAinsu30=read.table("../data/mol_overlap/APA2molNuclear/NucAPAqtlsPval4su30.txt", header = T, stringsAsFactors = F)
qval_su30N=pi0est(NucAPAinsu30$pval, pi0.method = "bootstrap")- 4su60
NucAPAinsu60=read.table("../data/mol_overlap/APA2molNuclear/NucAPAqtlsPval4su60.txt", header = T, stringsAsFactors = F)
qval_su60N=pi0est(NucAPAinsu60$pval, pi0.method = "bootstrap")All plots:
par(mfrow=c(2,3))
hist(NucAPAinsu30$pval, xlab="4su30 Pvalue", main="Significant nuclear APA QTLs \n 4su30")
text(.6,80, paste("pi_1=", round((1-qval_su30N$pi0), digit=3), sep=" "))
hist(NucAPAinsu60$pval,xlab="4su60 Pvalue",main="Significant nuclear APA QTLs \n 4su60")
text(.6,90, paste("pi_1=", round((1-qval_su60N$pi0), digit=3), sep=" "))
hist(NucAPAinRNA$pval, xlab="RNA Pvalue",main="Significant nuclear APA QTLs \n RNA")
text(.6,100, paste("pi_1=", round((1-qval_RNAN$pi0), digit=3), sep=" "))
hist(NucAPAinRNAg$pval, xlab="RNA Guevadis Pvalue",main="Significant nuclear APA QTLs \n RNA Geuvadis")
text(.6,100, paste("pi_1=", round((1-qval_RNAgN$pi0), digit=3), sep=" "))
hist(NucAPAinRibo$pval, xlab="Ribo (translation) Pvalue",main="Significant nuclear APA QTLs \n Ribo")
text(.6,100, paste("pi_1=", round((1-qval_RiboN$pi0), digit=3), sep=" "))
hist(NucAPAinProt$pval, xlab="Protein Pvalue", main="Significant nuclear APA QTLs \n Protein")
text(.6,40, paste("pi_1=", round((1-qval_protN$pi0), digit=3), sep=" "))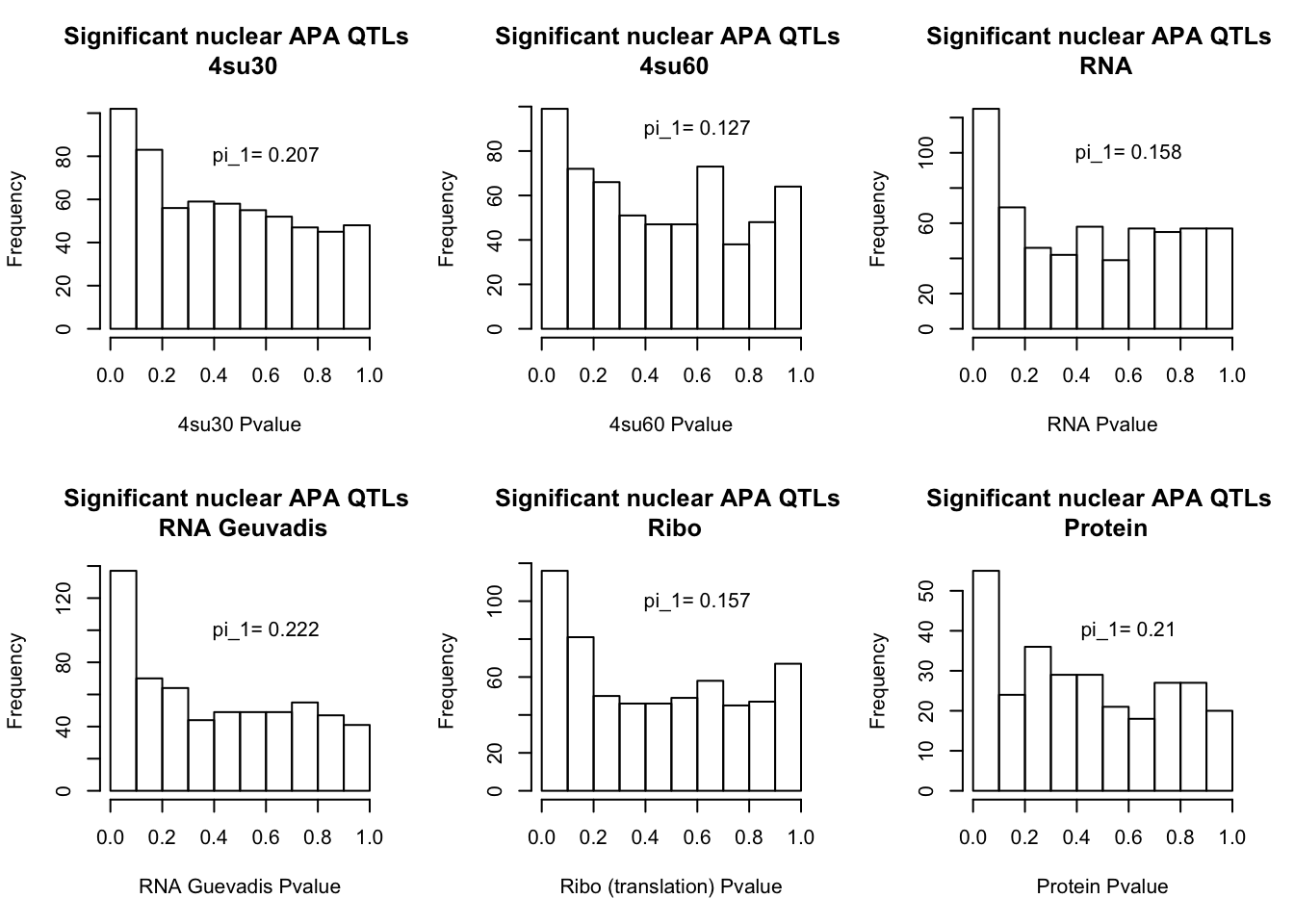
Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] qvalue_2.12.0 data.table_1.11.8 VennDiagram_1.6.20
[4] futile.logger_1.4.3 forcats_0.3.0 stringr_1.3.1
[7] dplyr_0.7.6 purrr_0.2.5 readr_1.1.1
[10] tidyr_0.8.1 tibble_1.4.2 ggplot2_3.0.0
[13] tidyverse_1.2.1 reshape2_1.4.3 workflowr_1.1.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.4 splines_3.5.1 haven_1.1.2
[4] lattice_0.20-35 colorspace_1.3-2 htmltools_0.3.6
[7] yaml_2.2.0 rlang_0.2.2 R.oo_1.22.0
[10] pillar_1.3.0 glue_1.3.0 withr_2.1.2
[13] R.utils_2.7.0 lambda.r_1.2.3 modelr_0.1.2
[16] readxl_1.1.0 bindrcpp_0.2.2 bindr_0.1.1
[19] plyr_1.8.4 munsell_0.5.0 gtable_0.2.0
[22] cellranger_1.1.0 rvest_0.3.2 R.methodsS3_1.7.1
[25] evaluate_0.11 knitr_1.20 broom_0.5.0
[28] Rcpp_0.12.19 formatR_1.5 backports_1.1.2
[31] scales_1.0.0 jsonlite_1.5 hms_0.4.2
[34] digest_0.6.17 stringi_1.2.4 rprojroot_1.3-2
[37] cli_1.0.1 tools_3.5.1 magrittr_1.5
[40] lazyeval_0.2.1 futile.options_1.0.1 crayon_1.3.4
[43] whisker_0.3-2 pkgconfig_2.0.2 xml2_1.2.0
[46] lubridate_1.7.4 assertthat_0.2.0 rmarkdown_1.10
[49] httr_1.3.1 rstudioapi_0.8 R6_2.3.0
[52] nlme_3.1-137 git2r_0.23.0 compiler_3.5.1
This reproducible R Markdown analysis was created with workflowr 1.1.1