ChromHMM analysis
Briana Mittleman
11/7/2018
Last updated: 2018-11-15
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: acf77f8
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: data/.DS_Store Ignored: output/.DS_Store Untracked files: Untracked: KalistoAbundance18486.txt Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed Untracked: analysis/snake.config.notes.Rmd Untracked: analysis/verifyBAM.Rmd Untracked: data/18486.genecov.txt Untracked: data/APApeaksYL.total.inbrain.bed Untracked: data/ChromHmmOverlap/ Untracked: data/GM12878.chromHMM.bed Untracked: data/GM12878.chromHMM.txt Untracked: data/NuclearApaQTLs.txt Untracked: data/PeaksUsed/ Untracked: data/RNAkalisto/ Untracked: data/TotalApaQTLs.txt Untracked: data/Totalpeaks_filtered_clean.bed Untracked: data/YL-SP-18486-T-combined-genecov.txt Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt Untracked: data/apaExamp/ Untracked: data/bedgraph_peaks/ Untracked: data/bin200.5.T.nuccov.bed Untracked: data/bin200.Anuccov.bed Untracked: data/bin200.nuccov.bed Untracked: data/clean_peaks/ Untracked: data/comb_map_stats.csv Untracked: data/comb_map_stats.xlsx Untracked: data/comb_map_stats_39ind.csv Untracked: data/combined_reads_mapped_three_prime_seq.csv Untracked: data/diff_iso_trans/ Untracked: data/ensemble_to_genename.txt Untracked: data/example_gene_peakQuant/ Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed Untracked: data/first50lines_closest.txt Untracked: data/gencov.test.csv Untracked: data/gencov.test.txt Untracked: data/gencov_zero.test.csv Untracked: data/gencov_zero.test.txt Untracked: data/gene_cov/ Untracked: data/joined Untracked: data/leafcutter/ Untracked: data/merged_combined_YL-SP-threeprimeseq.bg Untracked: data/mol_overlap/ Untracked: data/mol_pheno/ Untracked: data/nom_QTL/ Untracked: data/nom_QTL_opp/ Untracked: data/nom_QTL_trans/ Untracked: data/nuc6up/ Untracked: data/other_qtls/ Untracked: data/peakPerRefSeqGene/ Untracked: data/perm_QTL/ Untracked: data/perm_QTL_opp/ Untracked: data/perm_QTL_trans/ Untracked: data/reads_mapped_three_prime_seq.csv Untracked: data/smash.cov.results.bed Untracked: data/smash.cov.results.csv Untracked: data/smash.cov.results.txt Untracked: data/smash_testregion/ Untracked: data/ssFC200.cov.bed Untracked: data/temp.file1 Untracked: data/temp.file2 Untracked: data/temp.gencov.test.txt Untracked: data/temp.gencov_zero.test.txt Untracked: output/picard/ Untracked: output/plots/ Untracked: output/qual.fig2.pdf Unstaged changes: Modified: analysis/28ind.peak.explore.Rmd Modified: analysis/39indQC.Rmd Modified: analysis/cleanupdtseq.internalpriming.Rmd Modified: analysis/coloc_apaQTLs_protQTLs.Rmd Modified: analysis/dif.iso.usage.leafcutter.Rmd Modified: analysis/diff_iso_pipeline.Rmd Modified: analysis/explore.filters.Rmd Modified: analysis/flash2mash.Rmd Modified: analysis/overlapMolQTL.Rmd Modified: analysis/overlap_qtls.Rmd Modified: analysis/peakOverlap_oppstrand.Rmd Modified: analysis/pheno.leaf.comb.Rmd Modified: analysis/swarmPlots_QTLs.Rmd Modified: analysis/test.max2.Rmd Modified: code/Snakefile
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | acf77f8 | Briana Mittleman | 2018-11-15 | add res from uniq analysis |
| html | 89fa7d6 | Briana Mittleman | 2018-11-13 | Build site. |
| Rmd | 595cdc0 | Briana Mittleman | 2018-11-13 | add code for uniq no sig permutation |
| html | 086fbcc | Briana Mittleman | 2018-11-13 | Build site. |
| Rmd | 1357eac | Briana Mittleman | 2018-11-13 | res from no sig analysis |
| html | ff8c969 | Briana Mittleman | 2018-11-12 | Build site. |
| Rmd | d08f3f9 | Briana Mittleman | 2018-11-12 | fix code for no sig permutations |
| html | 83f1b14 | Briana Mittleman | 2018-11-08 | Build site. |
| Rmd | ab79f33 | Briana Mittleman | 2018-11-08 | add enrichment analysis (messed up perm) |
| html | 19b98b3 | Briana Mittleman | 2018-11-07 | Build site. |
| Rmd | 70cf09c | Briana Mittleman | 2018-11-07 | add perm res |
| html | 2ec5ffd | Briana Mittleman | 2018-11-07 | Build site. |
| Rmd | 962e39b | Briana Mittleman | 2018-11-07 | move chromhmm analysis to its own analysis |
Librarys
library(workflowr)This is workflowr version 1.1.1
Run ?workflowr for help getting startedlibrary(reshape2)
library(tidyverse)── Attaching packages ─────────────────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.0.0 ✔ purrr 0.2.5
✔ tibble 1.4.2 ✔ dplyr 0.7.6
✔ tidyr 0.8.1 ✔ stringr 1.3.1
✔ readr 1.1.1 ✔ forcats 0.3.0── Conflicts ────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(VennDiagram)Loading required package: gridLoading required package: futile.loggerlibrary(data.table)
Attaching package: 'data.table'The following objects are masked from 'package:dplyr':
between, first, lastThe following object is masked from 'package:purrr':
transposeThe following objects are masked from 'package:reshape2':
dcast, meltlibrary(ggpubr)Loading required package: magrittr
Attaching package: 'magrittr'The following object is masked from 'package:purrr':
set_namesThe following object is masked from 'package:tidyr':
extract
Attaching package: 'ggpubr'The following object is masked from 'package:VennDiagram':
rotatelibrary(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggpubr':
get_legendThe following object is masked from 'package:ggplot2':
ggsaveI am continuing the analysis I started in the characterization of the APAqtl analysis. I need to run permutations to enrichment statistics.
I created the significant SNP files in the Characterize Total APAqtl analysis analysis.
chromHmm=read.table("../data/ChromHmmOverlap/chromHMM_regions.txt", col.names = c("number", "name"), stringsAsFactors = F)
NuclearOverlapHMM=read.table("../data/ChromHmmOverlap/Nuc_overlapHMM.bed", col.names=c("chrom", "start", "end", "sid", "significance", "strand", "number"))
NuclearOverlapHMM$number=as.integer(NuclearOverlapHMM$number)
NuclearOverlapHMM_names=NuclearOverlapHMM %>% left_join(chromHmm, by="number")NuclearOverlapHMM_names$number=as.character(NuclearOverlapHMM_names$number)
ggplot(NuclearOverlapHMM_names, aes(x=number, fill=name)) + geom_bar() + labs(title="ChromHMM labels for Nuclear APAQtls" , y="Number of SNPs", x="Region")+theme(axis.text.x = element_text(angle = 90, hjust = 1))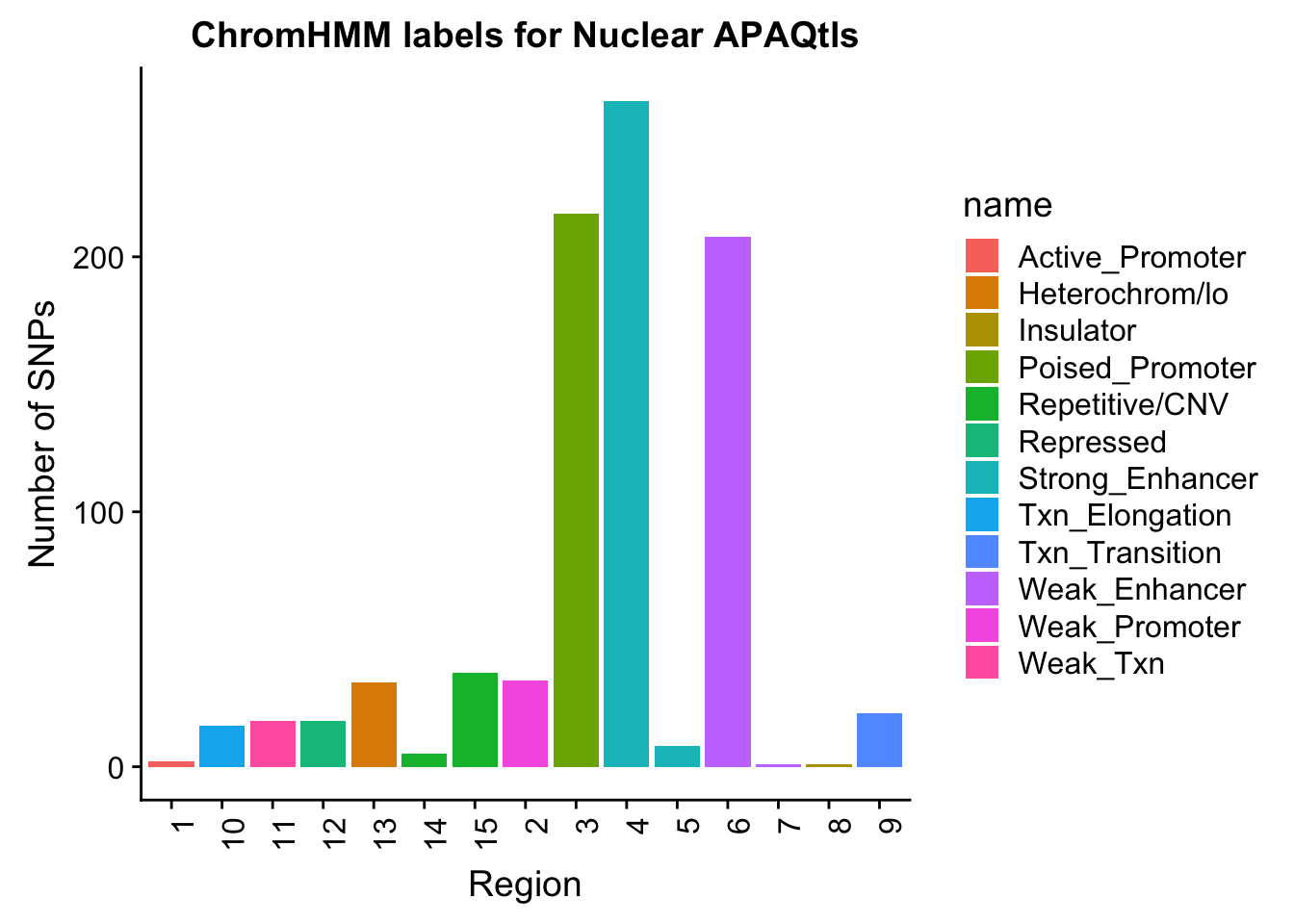
Expand here to see past versions of unnamed-chunk-3-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
Evaluate results for total:
TotalOverlapHMM=read.table("../data/ChromHmmOverlap/Tot_overlapHMM.bed", col.names=c("chrom", "start", "end", "sid", "significance", "strand", "number"))
TotalOverlapHMM_names=TotalOverlapHMM %>% left_join(chromHmm, by="number")TotalOverlapHMM_names$number=as.character(TotalOverlapHMM_names$number)
ggplot(TotalOverlapHMM_names, aes(x=number, fill=name)) + geom_bar() + labs(title="ChromHMM labels for Total APAQtls" , y="Number of SNPs", x="Region")+theme(axis.text.x = element_text(angle = 90, hjust = 1))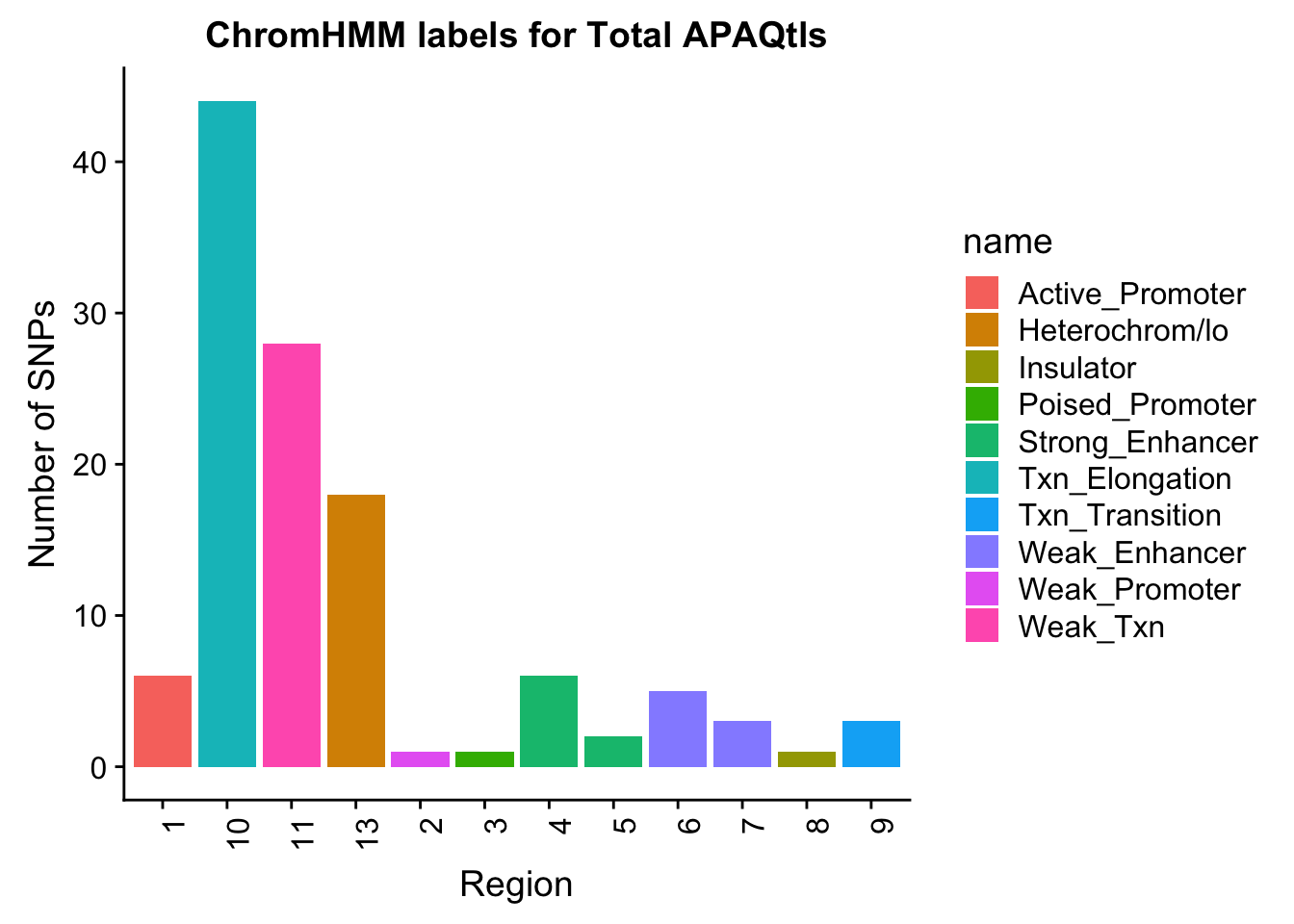
Expand here to see past versions of unnamed-chunk-5-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
Pull one set of random snps:
I do still need to get 880 random snps.
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/randomSnps/ApaQTL_nuclear_Random880.txt
Run QTLNOMres2SigSNPbed.py with nuclear 880 and sort output
import pybedtools
RANDnuc=pybedtools.BedTool('/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/randomSnps/ApaQTL_nuclear_Random880.sort.bed')
hmm=pybedtools.BedTool("/project2/gilad/briana/genome_anotation_data/GM12878.chromHMM.sort.bed")
#map hmm to snps
NucRnad_overlapHMM=RANDnuc.map(hmm, c=4)
#save results
NucRnad_overlapHMM.saveas("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/randomSnps/ApaQTL_nuclear_Random_overlapHMM.bed")
NuclearRandOverlapHMM=read.table("../data/ChromHmmOverlap/ApaQTL_nuclear_Random_overlapHMM.bed", col.names=c("chrom", "start", "end", "sid", "significance", "strand", "number"))
NuclearRandOverlapHMM_names=NuclearRandOverlapHMM %>% left_join(chromHmm, by="number")ggplot(NuclearRandOverlapHMM_names, aes(x=name)) + geom_bar() + labs(title="ChromHMM labels for Nuclear APAQtls (Random)" , y="Number of SNPs", x="Region")+theme(axis.text.x = element_text(angle = 90, hjust = 1))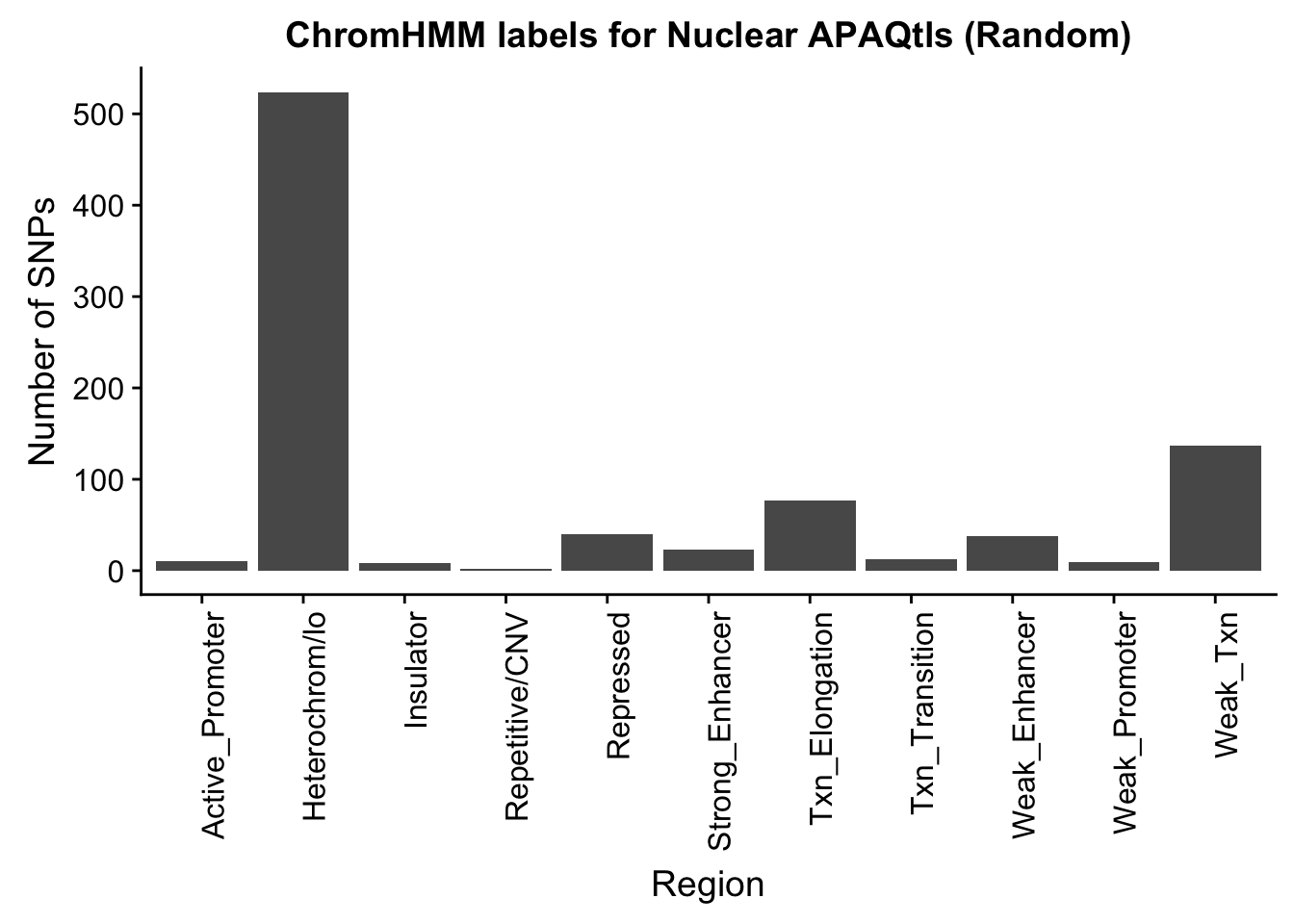
Expand here to see past versions of unnamed-chunk-9-1.png:
| Version | Author | Date |
|---|---|---|
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
To put this on the same plot I can count the number in each then plot them next to eachother.
random_perChromHMM_nuc=NuclearRandOverlapHMM_names %>% group_by(name) %>% summarise(Random=n())
sig_perChromHMM_nuc= NuclearOverlapHMM_names %>% group_by(name) %>% summarise(Nuclear_QTLs=n())
perChrommHMM_nuc=random_perChromHMM_nuc %>% full_join(sig_perChromHMM_nuc, by="name", ) %>% replace_na(list(Random=0,Total_QTLs=0))
perChrommHMM_nuc_melt=melt(perChrommHMM_nuc, id.vars="name")
names(perChrommHMM_nuc_melt)=c("Region","Set", "N_Snps" )chromenrichNuclearplot=ggplot(perChrommHMM_nuc_melt, aes(x=Region, y=N_Snps, by=Set, fill=Set)) + geom_bar(position="dodge", stat="identity") +theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(title="Enrichment of Nuclear QTLs by chromatin region", y="Number of Snps", x="Chromatin Region") + scale_fill_brewer(palette="Paired")
chromenrichNuclearplot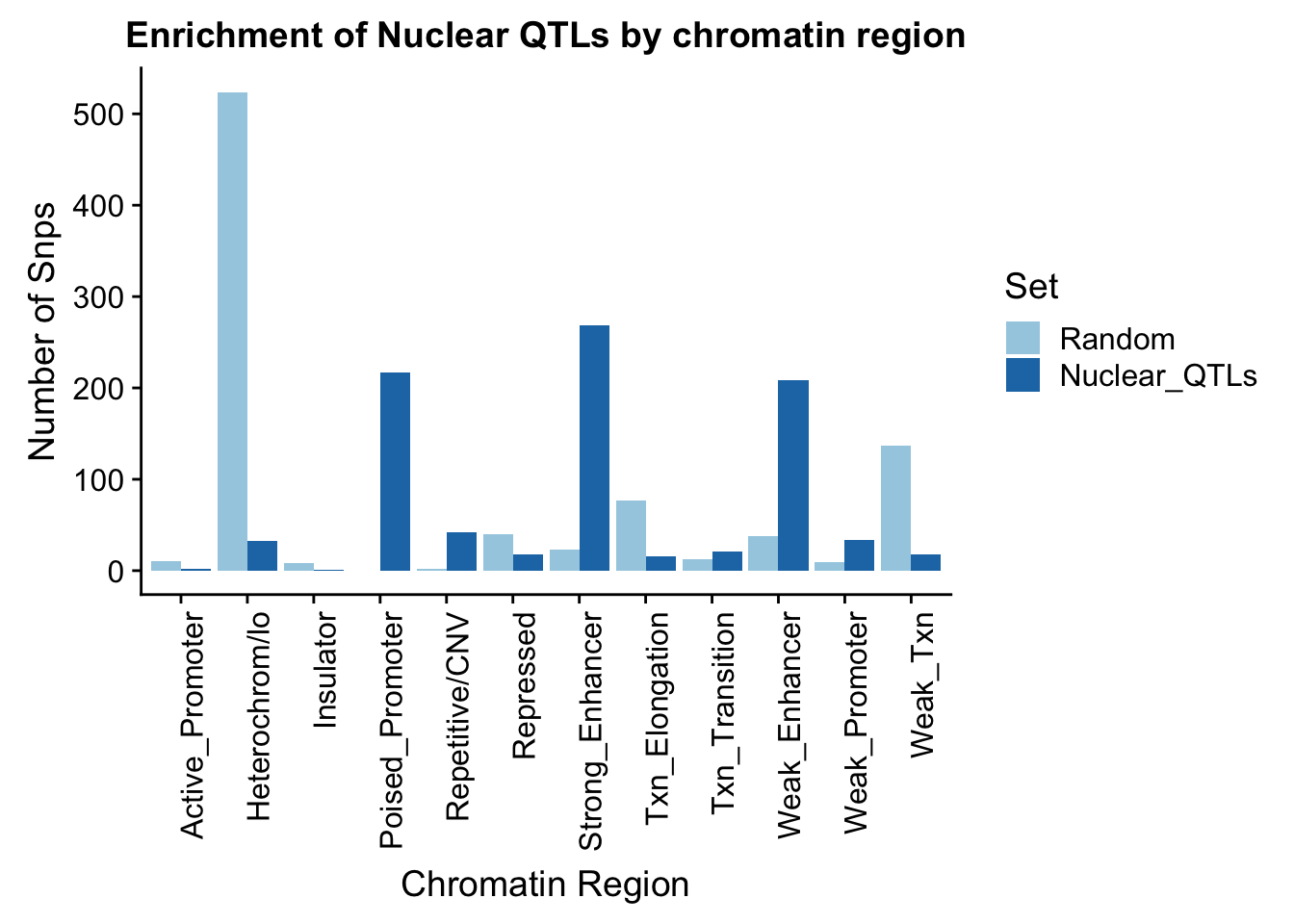
Expand here to see past versions of unnamed-chunk-11-1.png:
| Version | Author | Date |
|---|---|---|
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
ggsave("../output/plots/ChromHmmEnrich_Nuclear.png", chromenrichNuclearplot)Saving 7 x 5 in imageCompare enrichment between fractions
I want to make a plot with the enrichment by fraction. I am first going to get an enrichemnt score for each bin naively by looking at the QTL/random in each category.
#perChrommHMM_nuc$Random= as.integer(perChrommHMM_nuc$Random)
#perChrommHMM_nuc_enr=perChrommHMM_nuc %>% mutate(Nuclear=Nuclear_QTLs-Random)
#perChrommHMM_tot_enr=read.table("../data/ChromHmmOverlap/perChrommHMM_Total_enr.txt",stringsAsFactors = F,header = T)#allenrich=perChrommHMM_tot_enr %>% inner_join(perChrommHMM_nuc_enr, by="name") %>% select(name, Total, Nuclear)
#allenrich_melt=melt(allenrich, id.vars="name")plot it
#chromenrichBoth=ggplot(allenrich_melt, aes(x=name, by=variable, y=value, fill=variable)) + geom_bar(stat="identity", position = "dodge") + theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(title="QTL-Random for each bin by fraction", y="Num QTL SNPs - Num Random SNPs") + scale_fill_manual(values=c("darkviolet", "deepskyblue3"))
#ggsave("../output/plots/ChromHmmEnrich_BothFrac.png", chromenrichBoth)Permutations
I want to permute the background snps so i can get a better expectation. To do this I need to chose random lines from the nominal file, change the lines to snp format, overlap with HMM, count how many are in each category, and append the list to a dataframe that is category by permuation.
DO this for total first (118 snps)
total_random118_chromHmm.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_f
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_f.out
#SBATCH --error=total_random118_chromHmm_f.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {1..1000};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/randomRes_Total_118_${i}.txt
done
#make random
for i in {1..1000};
do
python randomRes2SNPbed.py Total 118 ${i}
done
#cat res together
cat /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed/* > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed_all/randomRes_Total_118_ALLperm.bed
#sort full file
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed_all/randomRes_Total_118_ALLperm.bed > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed_all/randomRes_Total_118_ALLperm.sort.bed
#hmm overlap
python overlap_chromHMM.py Total 118 1000
#Next I would pull this into R to do the group by and average!
pull_random_lines.py
def main(inFile, outFile ,nsamp):
nom_res= pd.read_csv(inFile, sep="\t", encoding="utf-8",header=None)
out=open(outFile, "w")
sample=nom_res.sample(nsamp)
sample.to_csv(out, sep="\t", encoding='utf-8', index=False, header=F)
out.close()
if __name__ == "__main__":
import sys
import pandas as pd
fraction = sys.argv[1]
nsamp=sys.argv[2]
nsamp=int(nsamp)
iter=sys.argv[3]
inFile = "/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_%s_NomRes.txt"%(fraction)
outFile = "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/randomRes_%s_%d_%s.txt"%(fraction,fraction, nsamp, iter)
main(inFile, outFile, nsamp)randomRes2SNPbed.py
def main(inFile, outFile):
fout=open(outFile, "w")
fin=open(inFile, "r")
for ln in fin:
pid, sid, dist, pval, slope = ln.split()
chrom, pos= sid.split(":")
name=sid
start= int(pos)-1
end=int(pos)
strand=pid.split(":")[3].split("_")[1]
pval=float(pval)
fout.write("%s\t%s\t%s\t%s\t%s\t%s\n"%(chrom, start, end, name, pval, strand))
fout.close()
if __name__ == "__main__":
import sys
fraction=sys.argv[1]
nsamp=sys.argv[2]
nsamp=int(nsamp)
iter=sys.argv[3]
inFile = "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/randomRes_%s_%d_%s.txt"%(fraction,fraction, nsamp, iter)
outFile= "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/snp_bed/randomRes_%s_%d_%s.bed"%(fraction,fraction, nsamp, iter)
main(inFile,outFile) overlap_chromHMM.py
def main(inFile, outFile):
rand=pybedtools.BedTool(inFile)
hmm=pybedtools.BedTool("/project2/gilad/briana/genome_anotation_data/GM12878.chromHMM.sort.bed")
#map hmm to snps
Rand_overlapHMM=rand.map(hmm, c=4)
#save results
Rand_overlapHMM.saveas(outFile)
if __name__ == "__main__":
import sys
import pandas as pd
import pybedtools
fraction=sys.argv[1]
nsamp=sys.argv[2]
niter=sys.argv[3]
inFile = "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/snp_bed_all/randomRes_%s_%s_ALLperm.sort.bed"%(fraction,fraction, nsamp)
outFile= "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/chromHMM_overlap/randomres_overlapChromHMM_%s_%s_%s.txt"%(fraction,fraction,nsamp, niter)
main(inFile,outFile)
*Nuclear 880
nuclear_random880_chromHmm.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=nuc_random880_chromHmm.out
#SBATCH --error=nuc_random880_chromHmm.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {1..1000};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/randomRes_Nuclear_880_${i}.txt
done
#make random
for i in {1..1000};
do
python randomRes2SNPbed.py Nuclear 880 ${i}
done
#cat res together
cat /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed/* > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_all/randomRes_Nuclear_880_ALLperm.bed
#sort full file
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_all/randomRes_Nuclear_880_ALLperm.bed > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_all/randomRes_Nuclear_880_ALLperm.sort.bed
#hmm overlap
python overlap_chromHMM.py Nuclear 880 1000
#Next I would pull this into R to do the group by and average!
Perm didnt finish: do this with less (824)
nuclear_random880_chromHmm.sm.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_sm
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_sm.out
#SBATCH --error=nuc_random880_chromHmm_sm.err
#SBATCH --partition=bigmem2
#SBATCH --mem=100G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {1..824};
do
python randomRes2SNPbed.py Nuclear 880 ${i}
done
#cat res together
cat /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed/* > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_all/randomRes_Nuclear_880_ALLperm.bed
#sort full file
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_all/randomRes_Nuclear_880_ALLperm.bed > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_all/randomRes_Nuclear_880_ALLperm.sort.bed
#hmm overlap
python overlap_chromHMM.py Nuclear 880 824I need a way to make this more efficient to run 1000 permutations. Here I will look at the results from the 824 permutations.
nuclear_perm824= read.table("../data/ChromHmmOverlap/randomres_overlapChromHMM_Nuclear_880_824.txt", col.names=c("chrom", "start", "end", "sid", "significance", "strand", "number"),stringsAsFactors = F, na.strings = "NA")
#924 snps are not annoated
nuclear_perm824$number=as.integer(as.factor(nuclear_perm824$number))
nuclear_perm824_names=nuclear_perm824 %>% left_join(chromHmm, by="number")
random_perChromHMM_nuc_PERM=nuclear_perm824_names %>% group_by(name) %>% summarise(Random=n()) %>% mutate(Random_perm=Random/824) %>% replace_na(list(name="No_annoation"))
perChrommHMM_nuc_withPerm=random_perChromHMM_nuc_PERM %>% full_join(sig_perChromHMM_nuc, by="name" ) %>% replace_na(list(Random=0,Nuclear_QTLs=0)) %>% select(name,Random_perm, Nuclear_QTLs)
perChrommHMM_nuc_withPerm_melt=melt(perChrommHMM_nuc_withPerm, id.vars="name")
names(perChrommHMM_nuc_withPerm_melt)=c("Region","Set", "N_Snps" )
ggplot(perChrommHMM_nuc_withPerm_melt, aes(x=Region, y=N_Snps, by=Set, fill=Set)) + geom_bar(position="dodge", stat="identity") +theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(title="Enrichment of Nuclear QTLs by chromatin region", y="Number of Snps", x="Chromatin Region") + scale_fill_brewer(palette="Paired")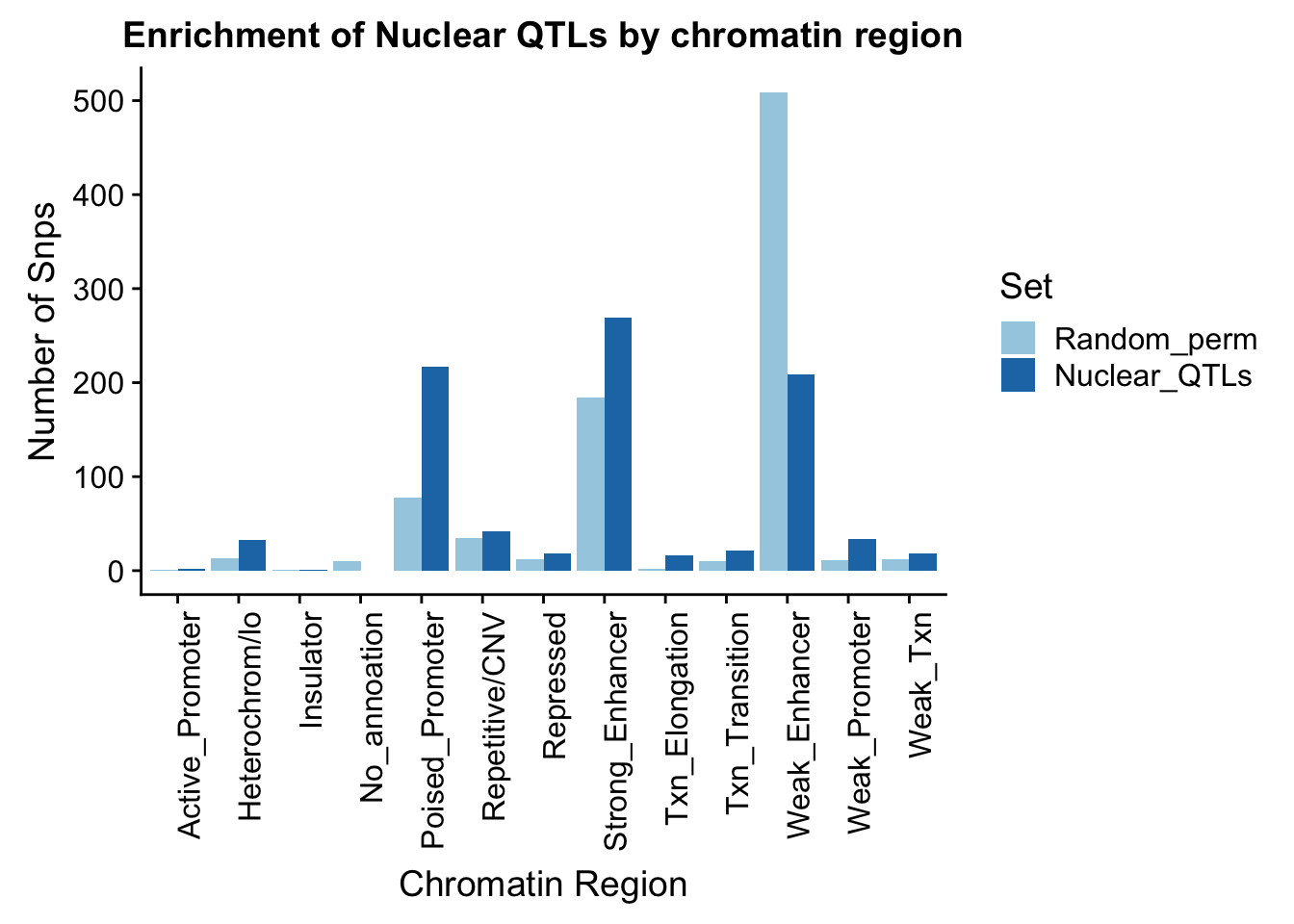
Expand here to see past versions of unnamed-chunk-21-1.png:
| Version | Author | Date |
|---|---|---|
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
Enrichment is the actual/random:
perChrommHMM_nuc_withPerm_enrich = perChrommHMM_nuc_withPerm %>% mutate(Nuclear_Enrichment=(Nuclear_QTLs-Random_perm)/Random_perm, chiSq=(Nuclear_QTLs-Random_perm)^2/Random_perm)
ggplot(perChrommHMM_nuc_withPerm_enrich, aes(x=name, y=Nuclear_Enrichment)) + geom_bar(stat="identity",fill="deepskyblue3")+ theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(title="ChromHMM Enrichment of Nuclear ApaQTLs \n over 824 Random Permuations", x="Region")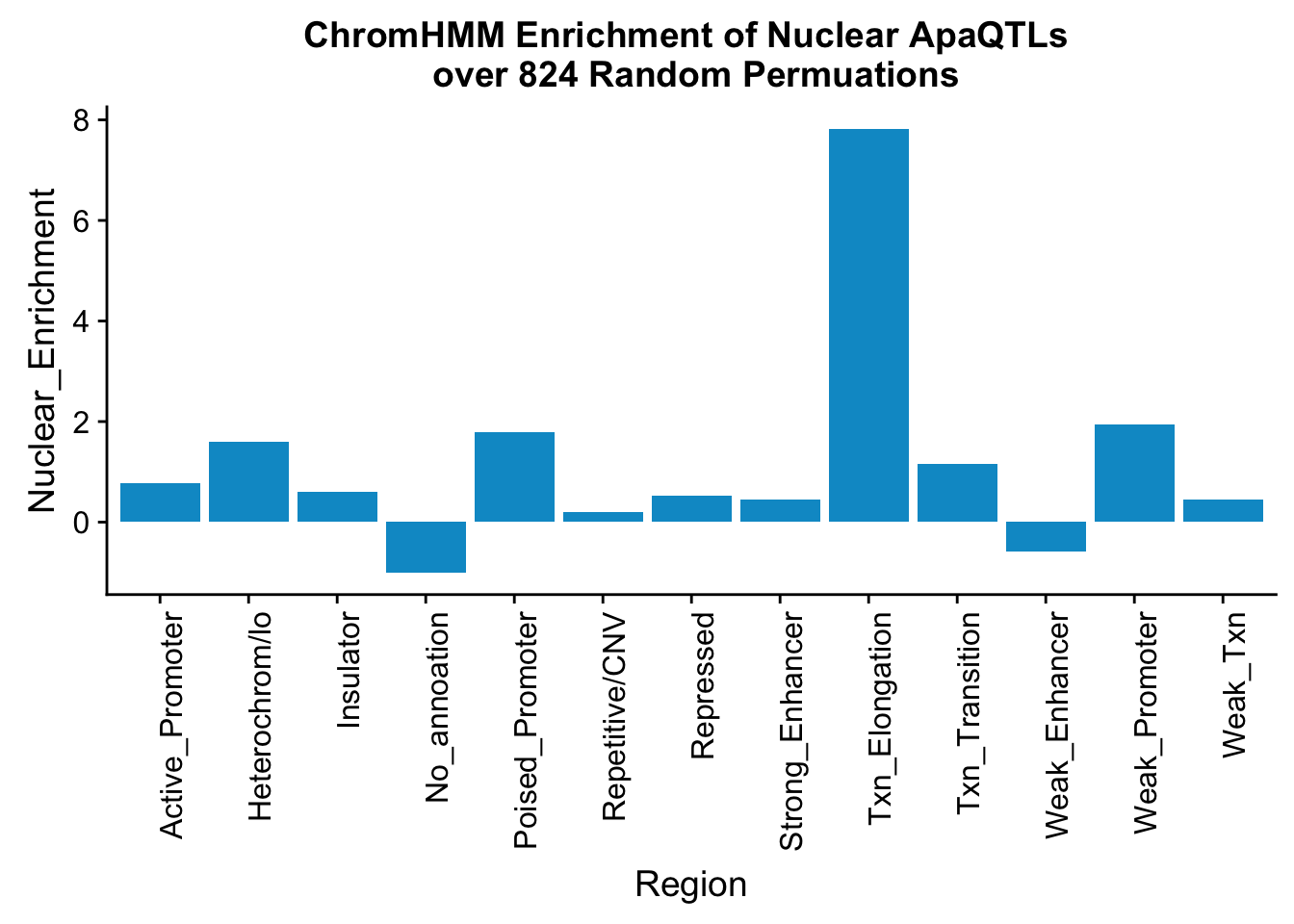
Expand here to see past versions of unnamed-chunk-22-1.png:
| Version | Author | Date |
|---|---|---|
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
ggplot(perChrommHMM_nuc_withPerm_enrich, aes(x=name, y=chiSq)) + geom_bar(stat="identity",fill="deepskyblue3")+ theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(title="ChromHMM ChiSq of Nuclear ApaQTLs \n over 824 Random Permuations", x="Region") 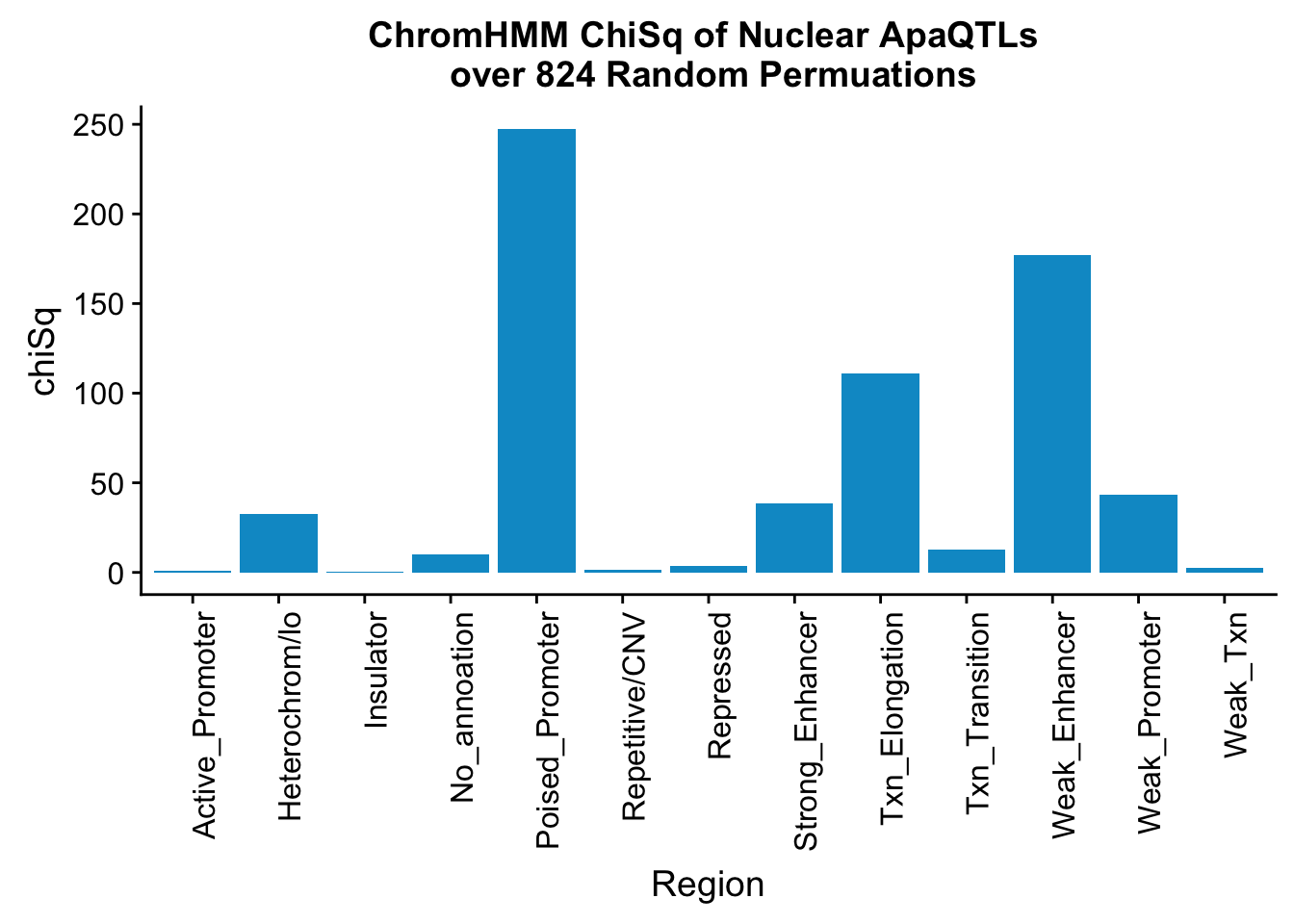
Expand here to see past versions of unnamed-chunk-22-2.png:
| Version | Author | Date |
|---|---|---|
| 2ec5ffd | Briana Mittleman | 2018-11-07 |
To parallelize this I will run the permutations in 4 bash scripts:
nuc_random880_chromHmm_set1.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_set1
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_set1.out
#SBATCH --error=nuc_random880_chromHmm_set1.err
#SBATCH --partition=bigmem2
#SBATCH --mem=100G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {1..250};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/randomRes_Nuclear_880_${i}.txt
done
nuc_random880_chromHmm_set2.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_set2
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_set2.out
#SBATCH --error=nuc_random880_chromHmm_set2.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {251..500};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/randomRes_Nuclear_880_${i}.txt
done
nuc_random880_chromHmm_set3.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_set3
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_set3.out
#SBATCH --error=nuc_random880_chromHmm_set3.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {501..750};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/randomRes_Nuclear_880_${i}.txt
done
nuc_random880_chromHmm_set4.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_set4
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_set4.out
#SBATCH --error=nuc_random880_chromHmm_set4.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {751..1000};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/randomRes_Nuclear_880_${i}.txt
done
Same for total:
total_random118_chromHmm_set1.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_set1
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_set1.out
#SBATCH --error=total_random118_chromHmm_set1.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {1..250};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/randomRes_Total_118_${i}.txt
done
total_random118_chromHmm_set2.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_set2
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_set2.out
#SBATCH --error=total_random118_chromHmm_set2.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {251..500};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/randomRes_Total_118_${i}.txt
done
total_random118_chromHmm_set4.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_set4
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_set4.out
#SBATCH --error=total_random118_chromHmm_set4.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {751..1000};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_NomRes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/randomRes_Total_118_${i}.txt
done
I want to turn each of these into snp files:
randomLines2Snp.sh
#!/bin/bash
#SBATCH --job-name=randomLines2Snp
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=randomLines2Snp.out
#SBATCH --error=randomLines2Snp.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {1..1000};
do
python randomRes2SNPbed.py Nuclear 880 ${i}
done
#make random
for i in {1..1000};
do
python randomRes2SNPbed.py Total 118 ${i}
done Next step is the overlap. I want this to run on each seperatly.
sortRandomSnps.sh
#!/bin/bash
#SBATCH --job-name=sortRandomSnps
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=sortRandomSnps.out
#SBATCH --error=sortRandomSnps.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in $(ls /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed/);
do
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed/$i > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/snp_bed_sort/$i.sort.bed
done
for i in $(ls /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed/);
do
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed/$i > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/snp_bed_sort/$i.sort.bed
done
Rewrite overlap with ChromHMM script to do it on each file seperatly.
overlap_chromHMM_sepfiles.py
def main(inFile, outFile):
rand=pybedtools.BedTool(inFile)
hmm=pybedtools.BedTool("/project2/gilad/briana/genome_anotation_data/GM12878.chromHMM.sort.bed")
#map hmm to snps
Rand_overlapHMM=rand.map(hmm, c=4)
#save results
Rand_overlapHMM.saveas(outFile)
if __name__ == "__main__":
import sys
import pandas as pd
import pybedtools
fraction=sys.argv[1]
nsamp=sys.argv[2]
niter=sys.argv[3]
#which itteration we are on
inFile ="/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/snp_bed_sort/randomRes_%s_%s_%s.bed.sort.bed"%(fraction,fraction, nsamp, iter)
outFile= "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s/chromHMM_overlap/randomres_overlapChromHMM_%s_%s_%s.txt"%(fraction,fraction,nsamp, niter)
main(inFile,outFile)overlap_chromHMM_sepfiles.sh
#!/bin/bash
#SBATCH --job-name=overlap_chromHMM_sepfiles
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=overlap_chromHMM_sepfiles.out
#SBATCH --error=overlap_chromHMM_sepfiles.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
python overlap_chromHMM_sepfiles.py Nuclear 880 $i
done
for i in {1..1000};
do
python overlap_chromHMM_sepfiles.py Total 118 $i
doneI will next make an R script that will take in each file and perform the groupby command to get the number of snps in each group.
groupRandomByChromHMM.R
#!/bin/rscripts
# usage: groupRandomByChromHMM.R -f infile -o outfile
#this file will take any of the itterations and output a file with chrom hmm number, name, numberof snps
library(optparse)
library(dplyr)
library(tidyr)
library(ggplot2)
library(readr)
option_list = list(
make_option(c("-f", "--file"), action="store", default=NA, type='character',
help="input coverage file"),
make_option(c("-o", "--output"), action="store", default=NA, type='character',
help="output file")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
#interrupt execution if no file is supplied
if (is.null(opt$file)){
print_help(opt_parser)
stop("Need input file", call.=FALSE)
}
if (is.null(opt$output)){
print_help(opt_parser)
stop("Need output file", call.=FALSE)
}
randomSNPS=read.table(opt$file, col.names=c("chrom", "start", "end", "sid", "significance", "strand", "number"),stringsAsFactors = F, na.strings = "NA")
hmm_names=read.table("/project2/gilad/briana/genome_anotation_data/chromHMM_regions.txt", col.names = c("number", "name"),stringsAsFactors=F)
randomSNPS$number=as.integer(as.factor(randomSNPS$number))
randomSNPS_names= randomSNPS %>% left_join(hmm_names, by="number")
#split the name of the file to get the iteration number
fileSplit=strsplit(opt$file, "/")[[1]][10]
iter.txt=strsplit(fileSplit, "_")[[1]][5]
iter=substr(iter.txt, 1, nchar(iter.txt)-4)
randomSNPS_names_grouped=randomSNPS_names %>% group_by(number) %>% summarise(!!iter:=n()) %>% replace_na(list(name="No_annotation")) %>% dplyr::select(number, !!iter)
hmm_names$number=as.character(hmm_names$number)
final=hmm_names %>% left_join(randomSNPS_names_grouped,by="number")
write.table(final,opt$output,quote=FALSE, col.names = T, row.names = F)groupRandomChromHMM.sh
#!/bin/bash
#SBATCH --job-name=groupRandomChromHMM
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=groupRandomChromHMM.out
#SBATCH --error=groupRandomChromHMM.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
Rscript groupRandomByChromHMM.R -f /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/chromHMM_overlap/randomres_overlapChromHMM_Nuclear_880_${i}.txt -o /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/chromHMM_overlap_group/randomres_overlapChromHMM_Nuclear_880_${i}_grouped.txt
done
for i in {1..1000};
do
Rscript groupRandomByChromHMM.R -f /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/chromHMM_overlap/randomres_overlapChromHMM_Total_118_${i}.txt -o /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/chromHMM_overlap_group/randomres_overlapChromHMM_Total_118_${i}_grouped.txt
doneOnce I have the results I will paste the third column of each file together
cut -d$' ' -f 1,2 randomres_overlapChromHMM_Nuclear_880_1_grouped.txt > Nuc_chromOverlap.txt
for i in {1..1000};
do
paste -d" " Nuc_chromOverlap.txt <(cut -d" " -f 3 randomres_overlapChromHMM_Nuclear_880_${i}_grouped.txt) > tmp
mv tmp Nuc_chromOverlap.txt
done
cut -d$' ' -f 1,2 randomres_overlapChromHMM_Total_118_99_grouped.txt> Tot_chromOverlap.txt
for i in {1..1000};
do
paste -d" " Tot_chromOverlap.txt <(cut -d" " -f 3 randomres_overlapChromHMM_Total_118_${i}_grouped.txt) > tmp
mv tmp Tot_chromOverlap.txt
done
There will be NAs in this file. I will turn them into 0s when I bring it in R.
Pull files onto computer:
/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear/chromHMM_overlap_group/Nuc_chromOverlap.txt /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total/chromHMM_overlap_group/Tot_chromOverlap.txt
regions=c('Txn_Elongation','Weak_Txn','Repressed','Heterochrom/lo','Repetitive/CNV1','Repetitive/CNV2','Active_Promoter','Weak_Promoter','Poised_Promoter','Strong_Enhancer1','Strong_Enhancer2','Weak_Enhancer1','Weak_Enhancer2','Insulator','Txn_Transition')
permutationResTotal=read.table("../data/ChromHmmOverlap/Tot_chromOverlap.txt", header=T, stringsAsFactors = F)
permutationResTotal[is.na(permutationResTotal)] <- 0
permutationResTotal= as_data_frame(permutationResTotal)
permutationResTotal_noName=permutationResTotal[,3:ncol(permutationResTotal)]
totRand_mean=rowMeans(permutationResTotal_noName)/1000
permutationResNuclear=read.table("../data/ChromHmmOverlap/Nuc_chromOverlap.txt",header = T,stringsAsFactors = F)
permutationResNuclear[is.na(permutationResNuclear)] <- 0
permutationResNuclear_noName=permutationResNuclear[,3:ncol(permutationResNuclear)]
nucRand_mean=rowMeans(permutationResNuclear_noName)/1000allRand_mean_df= data.frame(cbind(regions,totRand_mean, nucRand_mean))
allRand_mean_df_melt=melt(allRand_mean_df, id.vars="regions")Warning: attributes are not identical across measure variables; they will
be droppedallRand_mean_df_melt$value= as.numeric(allRand_mean_df_melt$value)
ggplot(allRand_mean_df_melt, aes(y=value, x=regions, by=variable, fill=variable))+ geom_histogram(stat="identity", position="dodge") + theme(axis.text.x = element_text(angle = 90, hjust = 1))Warning: Ignoring unknown parameters: binwidth, bins, pad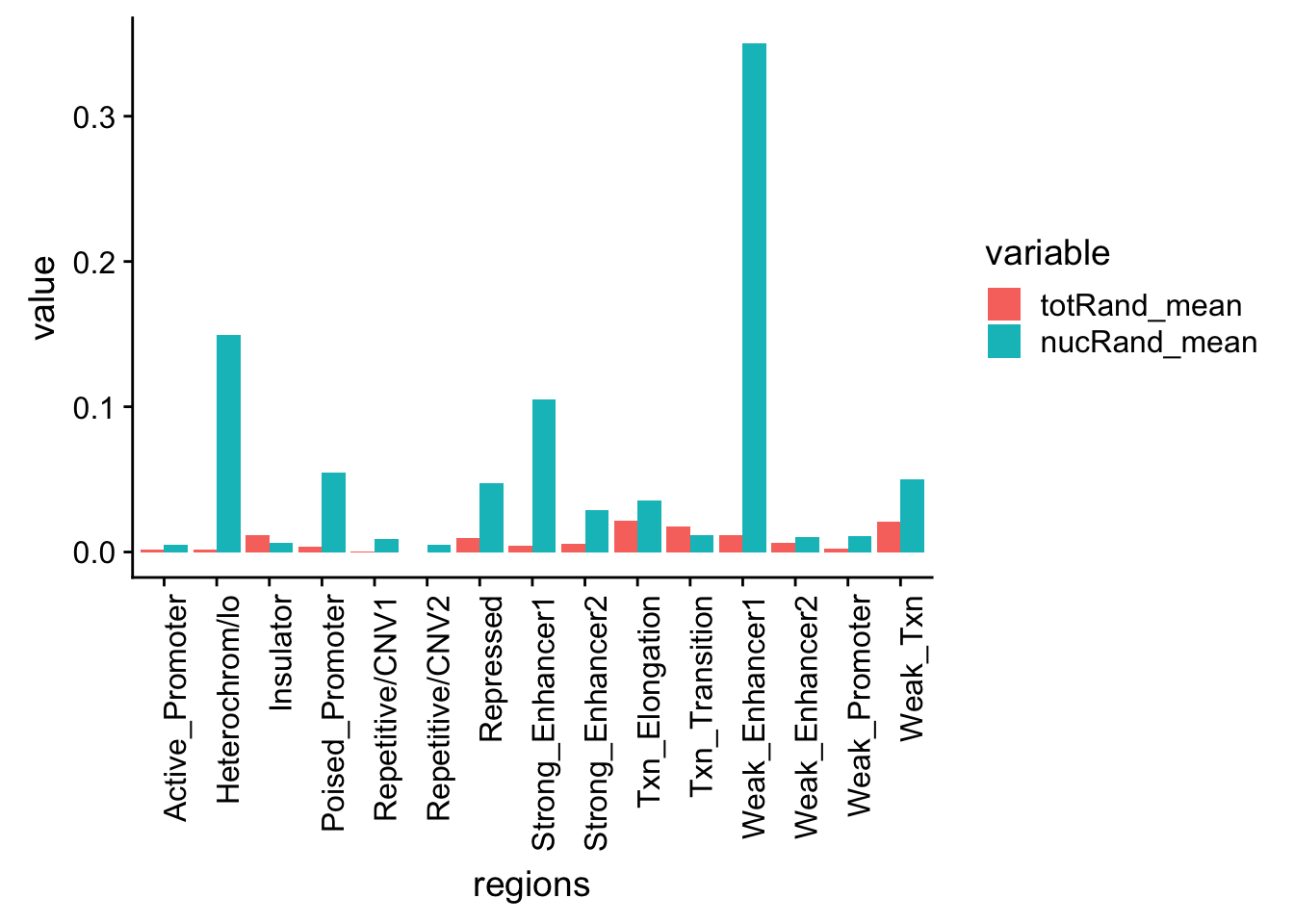
Expand here to see past versions of unnamed-chunk-38-1.png:
| Version | Author | Date |
|---|---|---|
| 19b98b3 | Briana Mittleman | 2018-11-07 |
I want to look at specific distributions:
permutationResTotal_melt= melt(permutationResTotal, id.vars=c("number", "name"))ggplot(permutationResTotal_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + labs(x="N random Snps in category", title="Random permutation Total")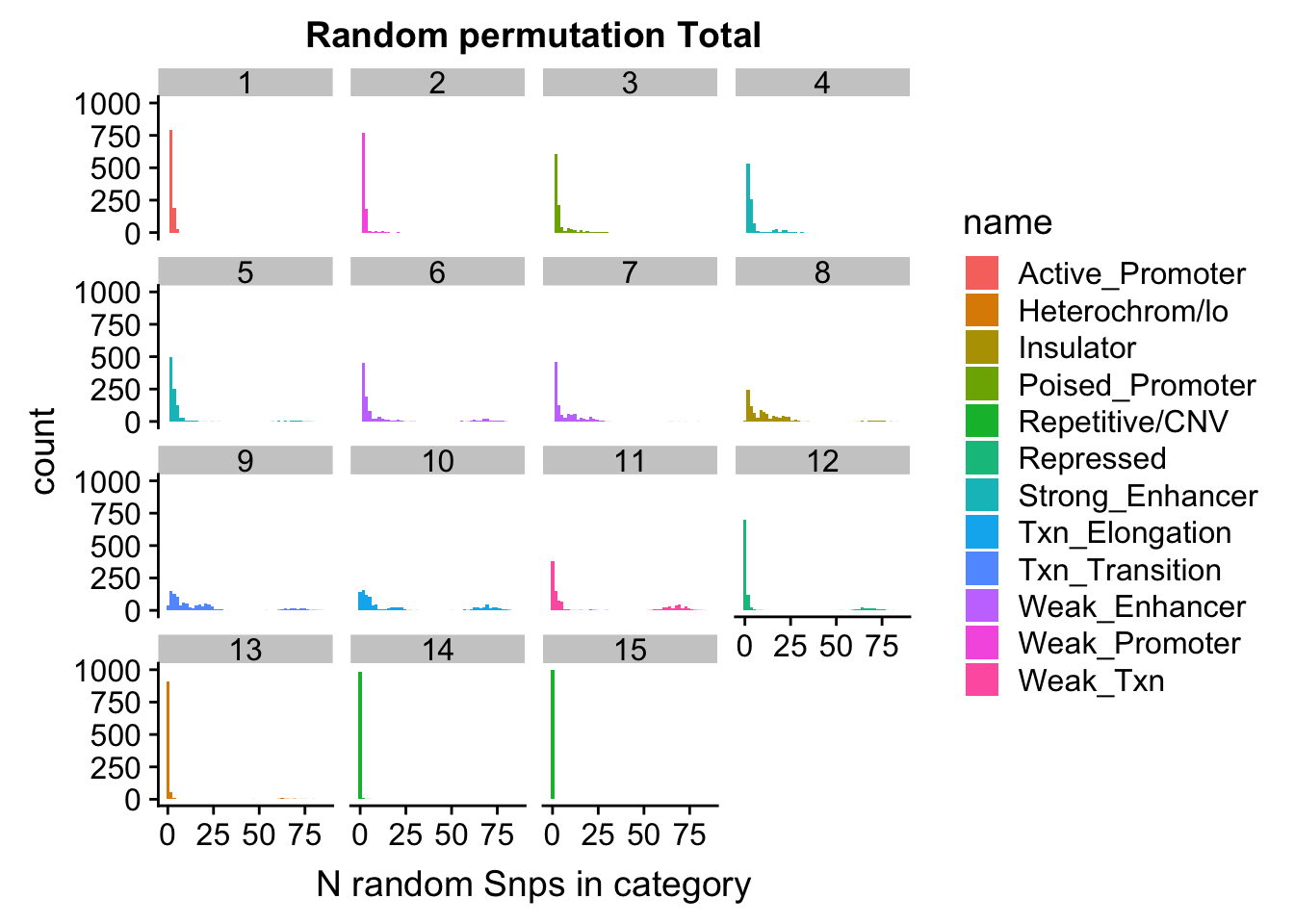
Expand here to see past versions of unnamed-chunk-40-1.png:
| Version | Author | Date |
|---|---|---|
| 19b98b3 | Briana Mittleman | 2018-11-07 |
For nuclear:
permutationResNuclear_melt= melt(permutationResNuclear, id.vars=c("number", "name"))ggplot(permutationResNuclear_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + labs(x="N random Snps in category", title="Random permutation Nuclear")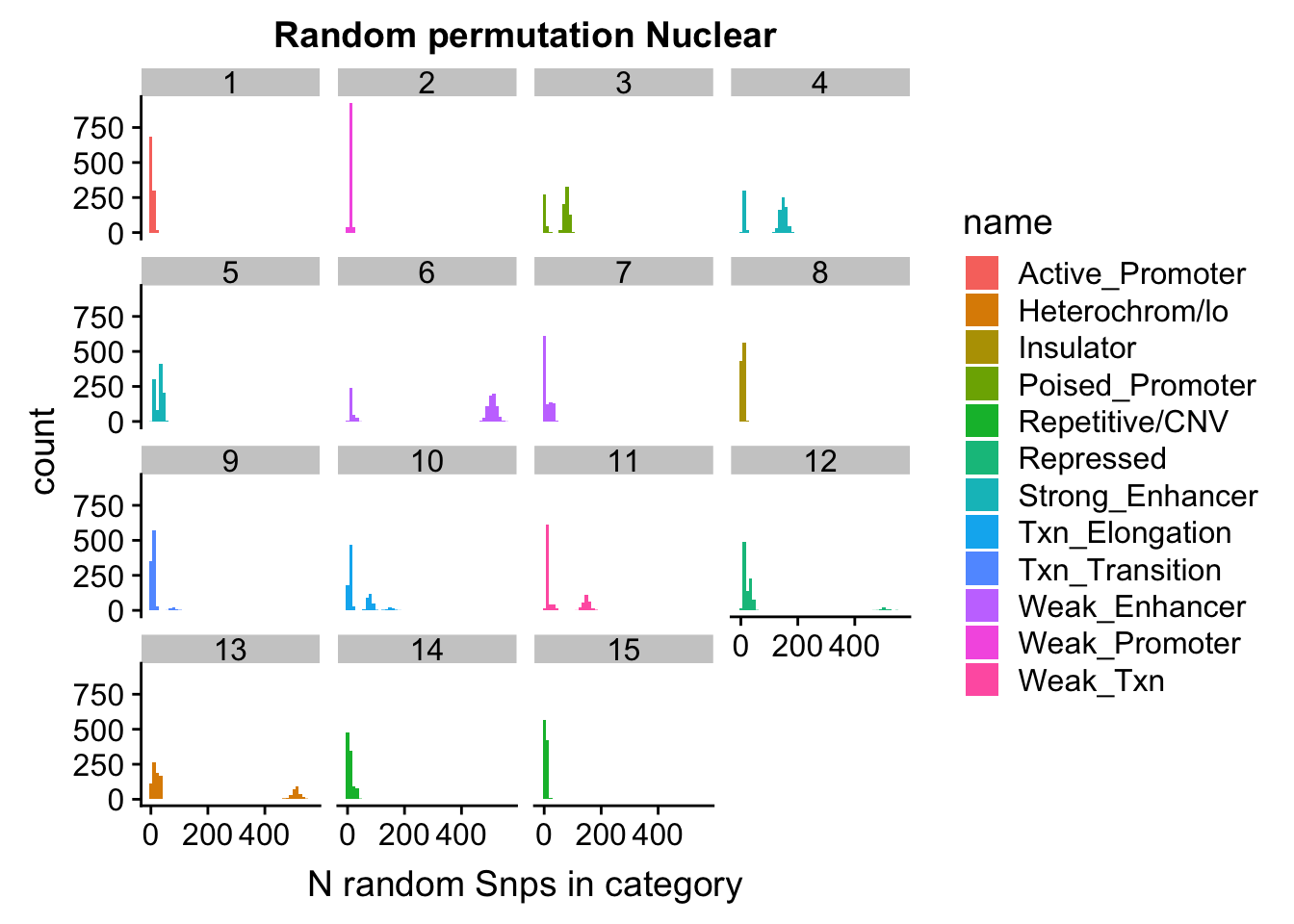
Expand here to see past versions of unnamed-chunk-42-1.png:
| Version | Author | Date |
|---|---|---|
| 19b98b3 | Briana Mittleman | 2018-11-07 |
Try log scale:
I want to add horizontal line where the actual QTL number is.
ggplot(permutationResTotal_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + scale_y_log10() + labs(x="random Snps in category", title="Random permutation Total")Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 448 rows containing missing values (geom_bar).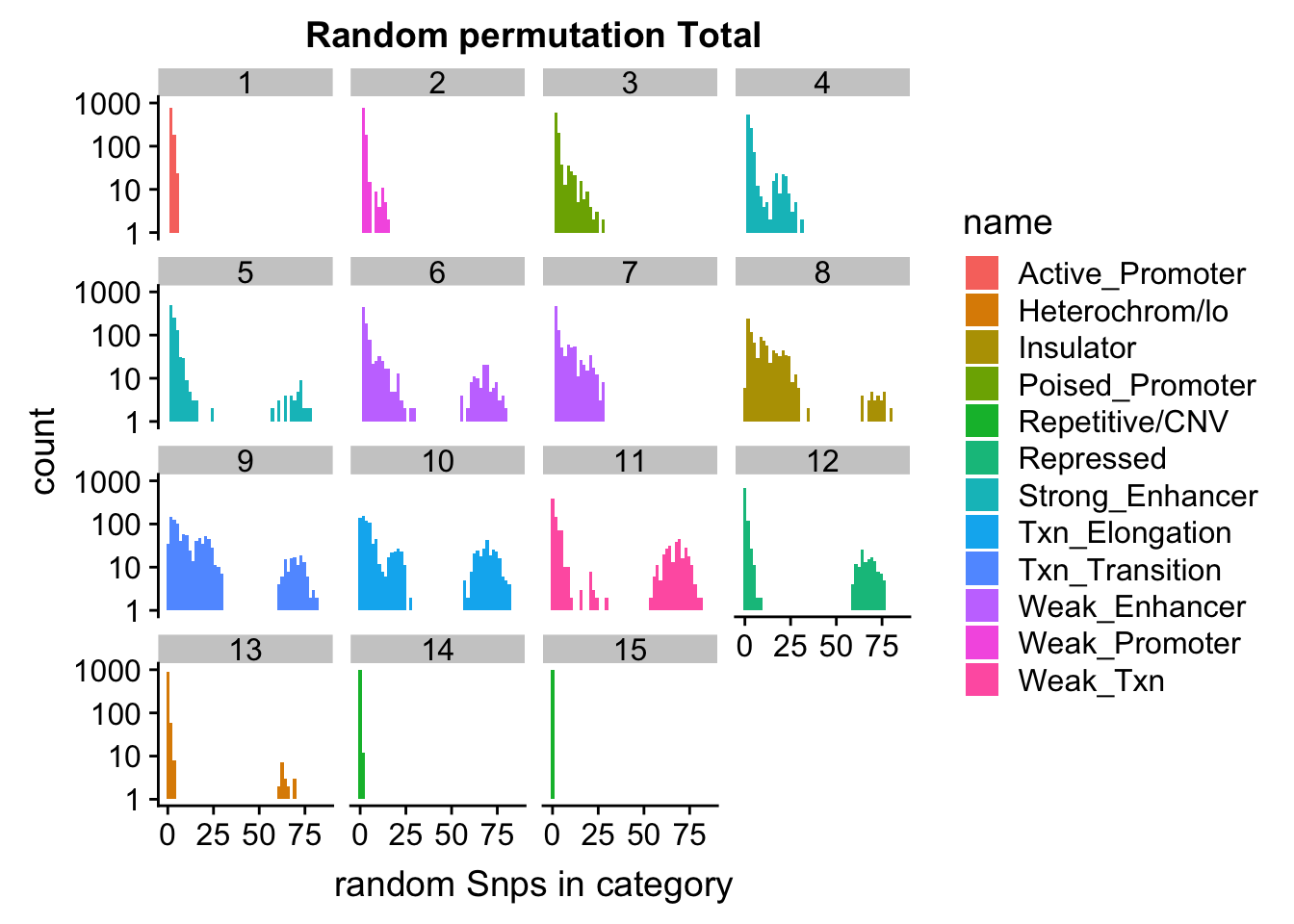
Expand here to see past versions of unnamed-chunk-43-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
| 19b98b3 | Briana Mittleman | 2018-11-07 |
ggplot(permutationResNuclear_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + scale_y_log10()+ labs(x="random Snps in category", title="Random permutation Nuclear")Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 631 rows containing missing values (geom_bar).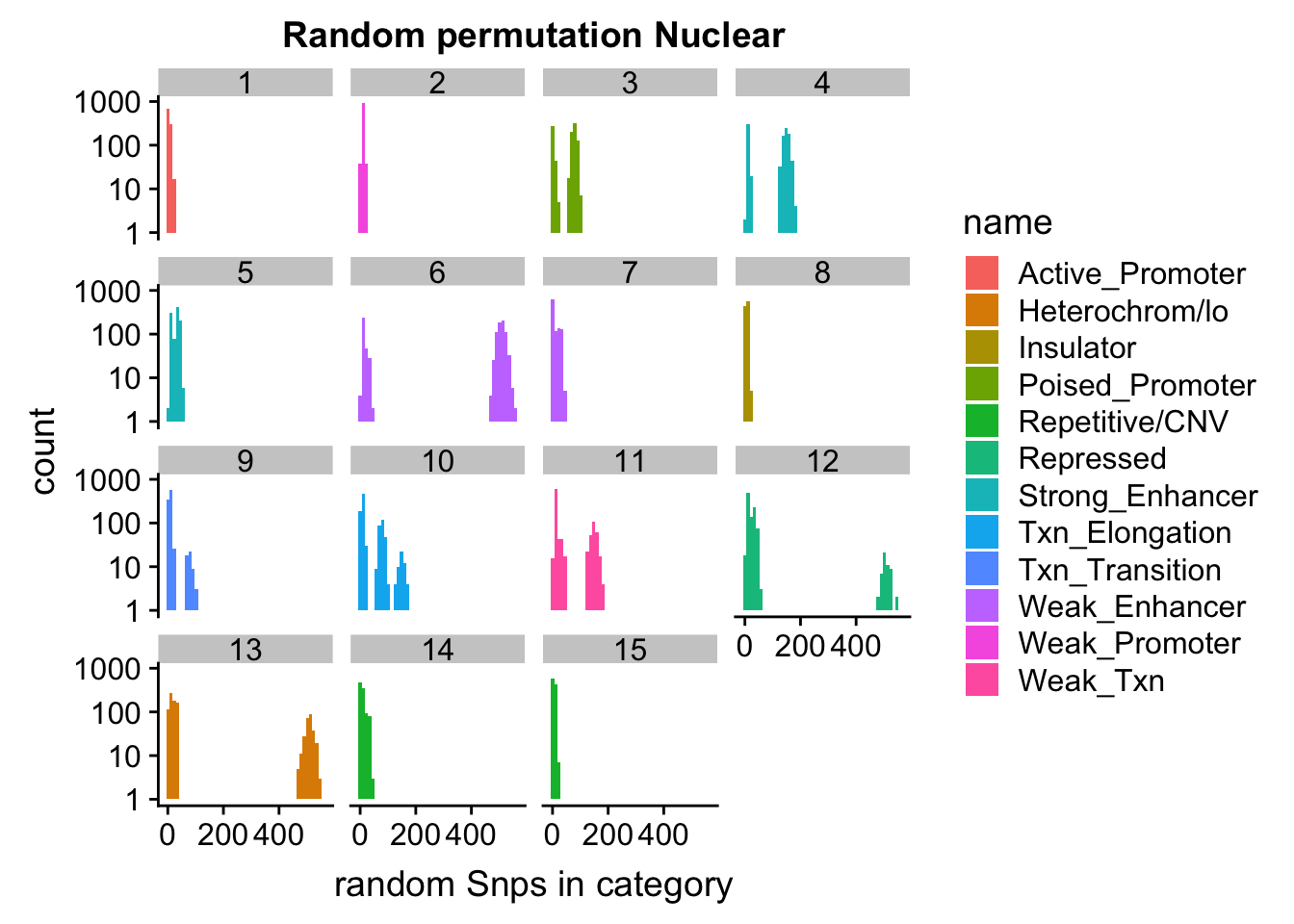
Expand here to see past versions of unnamed-chunk-44-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
| 19b98b3 | Briana Mittleman | 2018-11-07 |
Try removing 0s:
permutationResTotal_melt_no0= permutationResTotal_melt %>% filter(value>0)
ggplot(permutationResTotal_melt_no0, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number)+ scale_y_log10()+ labs(x="random Snps in category", title="Random permutation Total")Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 407 rows containing missing values (geom_bar).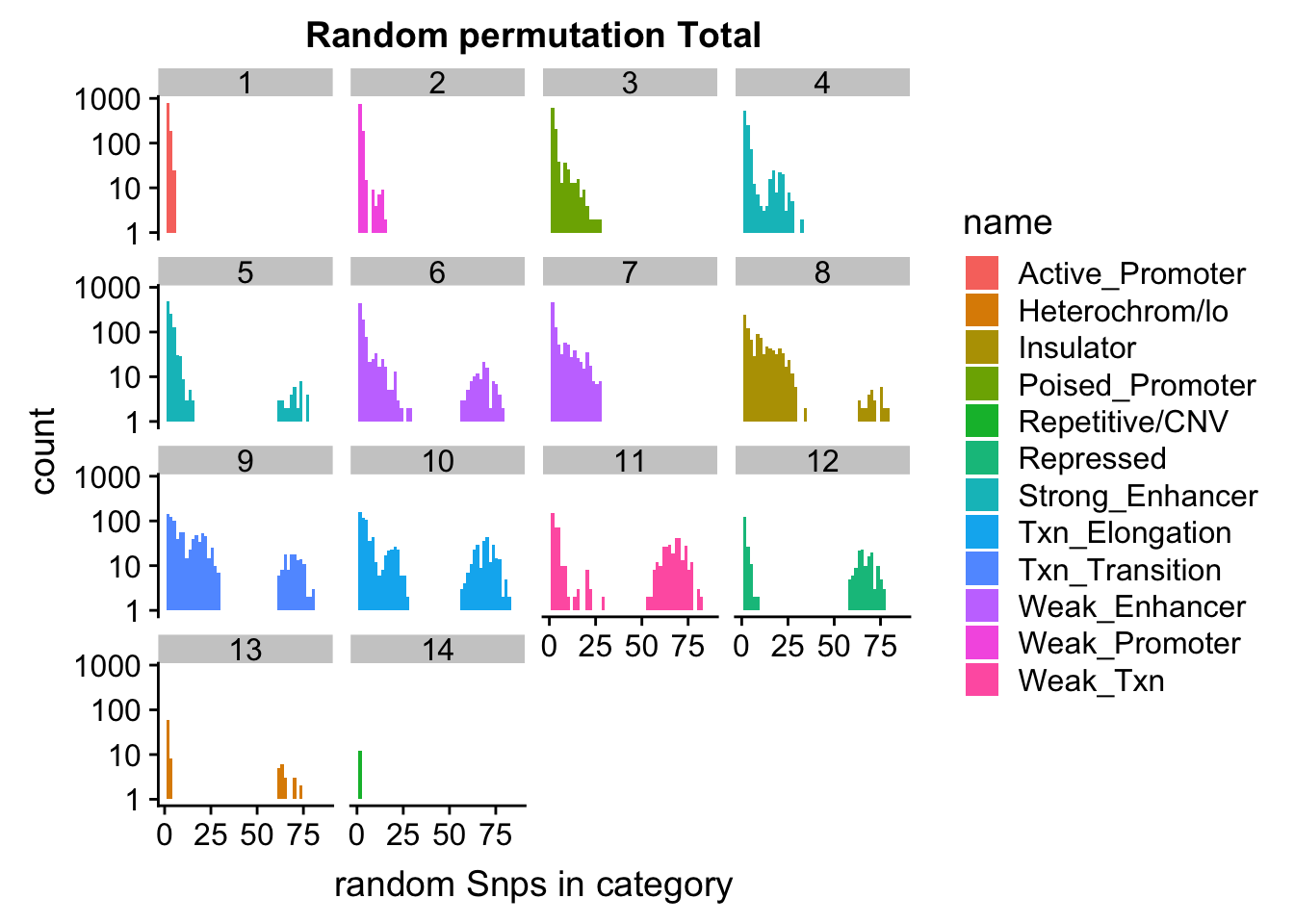
Expand here to see past versions of unnamed-chunk-45-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
| 19b98b3 | Briana Mittleman | 2018-11-07 |
permutationResNuclear_melt_no0= permutationResNuclear_melt %>% filter(value>0)
ggplot(permutationResNuclear_melt_no0, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number)+ scale_y_log10()+ labs(x="random Snps in category", title="Random permutation Nuclear")Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 630 rows containing missing values (geom_bar).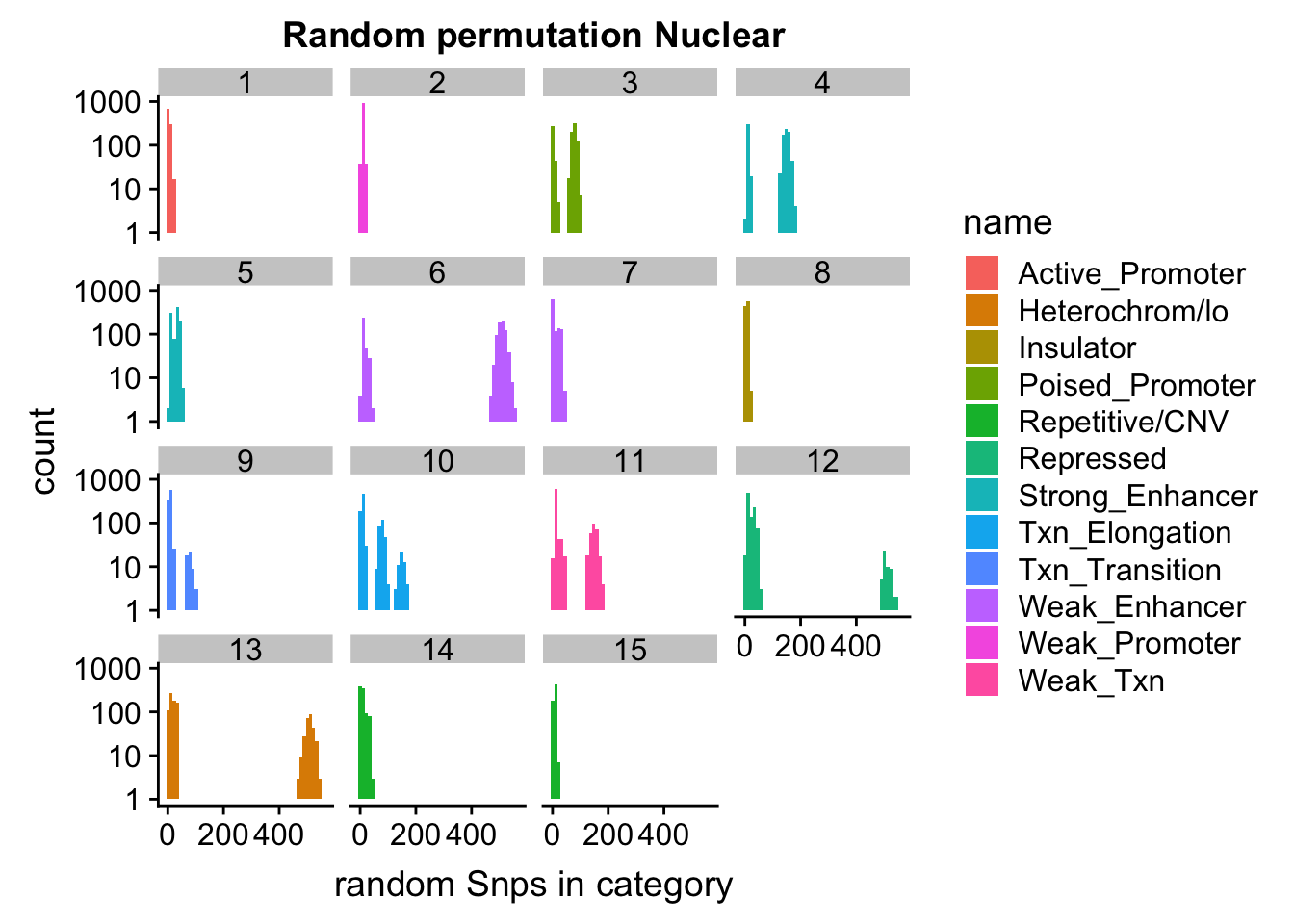
Expand here to see past versions of unnamed-chunk-45-2.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
| 19b98b3 | Briana Mittleman | 2018-11-07 |
Look at enrichment by using the average
TotalPermMean=permutationResTotal_melt %>% group_by(number) %>% summarise(TotRandPerm=mean(value))
TotalPermMean$number=as.character(TotalPermMean$number)
NuclearPermMean=permutationResNuclear_melt %>% group_by(number) %>% summarise(NucRandPerm=mean(value))
NuclearPermMean$number=as.character(NuclearPermMean$number)Melt SNP values by name and number to get data in same format:
TotalOverlapHMM_names_melt=melt(TotalOverlapHMM_names, id.vars=c("number", "name"))%>% filter(variable=="sid") %>% group_by(number) %>% summarise(TotalQTL=n())Warning: attributes are not identical across measure variables; they will
be droppedTotalOverlapHMM_names_melt$number=as.character(TotalOverlapHMM_names_melt$number)
NuclearOverlapHMM_names_melt=melt(NuclearOverlapHMM_names, id.vars=c("number", "name")) %>% filter(variable=="sid") %>% group_by(number) %>% summarise(NucQTL=n())Warning: attributes are not identical across measure variables; they will
be droppedNuclearOverlapHMM_names_melt$number=as.character(NuclearOverlapHMM_names_melt$number)chromHmm$number=as.character(chromHmm$number)
TotalOverlapHMM_enrichment= TotalOverlapHMM_names_melt %>% full_join(TotalPermMean, by="number") %>% replace_na(list(TotalQTL=.00001)) %>% full_join(chromHmm, by="number")
TotalOverlapHMM_enrichment$TotalQTL=as.double(TotalOverlapHMM_enrichment$TotalQTL)
TotalOverlapHMM_enrichment = TotalOverlapHMM_enrichment %>% mutate(TotEnrich=(TotalQTL-TotRandPerm)/TotRandPerm)
NuclearOverlapHMM_enrichment=NuclearOverlapHMM_names_melt %>% full_join(NuclearPermMean, by="number")%>% full_join(chromHmm, by="number")
NuclearOverlapHMM_enrichment$NucQTL=as.double(NuclearOverlapHMM_enrichment$NucQTL)
NuclearOverlapHMM_enrichment=NuclearOverlapHMM_enrichment %>%mutate(NucEnrich=(NucQTL-NucRandPerm)/NucRandPerm)ggplot(NuclearOverlapHMM_enrichment, aes(y=NucEnrich, x=number, fill=name)) + geom_bar(stat="identity")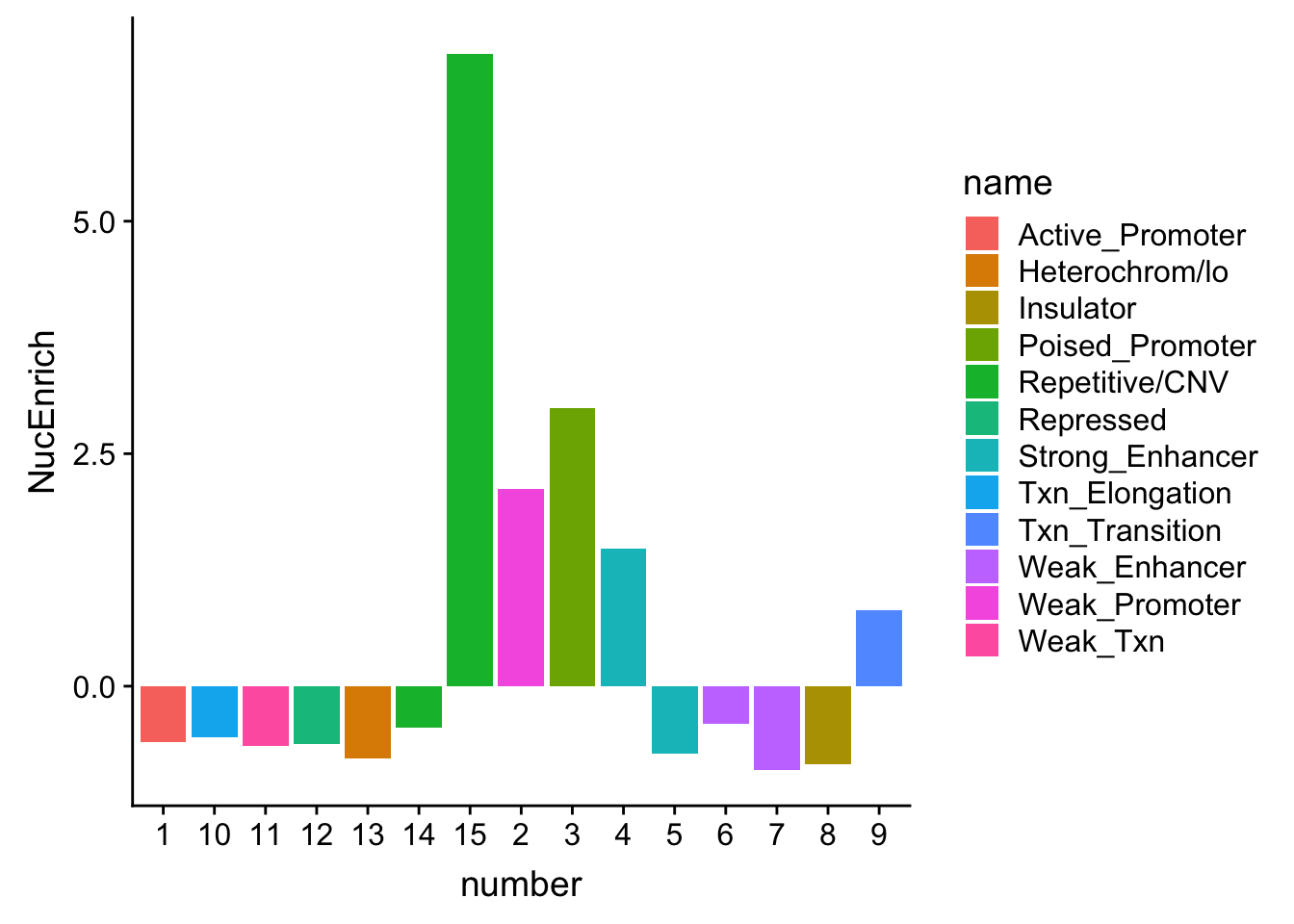
Expand here to see past versions of unnamed-chunk-49-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
ggplot(TotalOverlapHMM_enrichment, aes(y=TotEnrich, x=number, fill=name)) + geom_bar(stat="identity")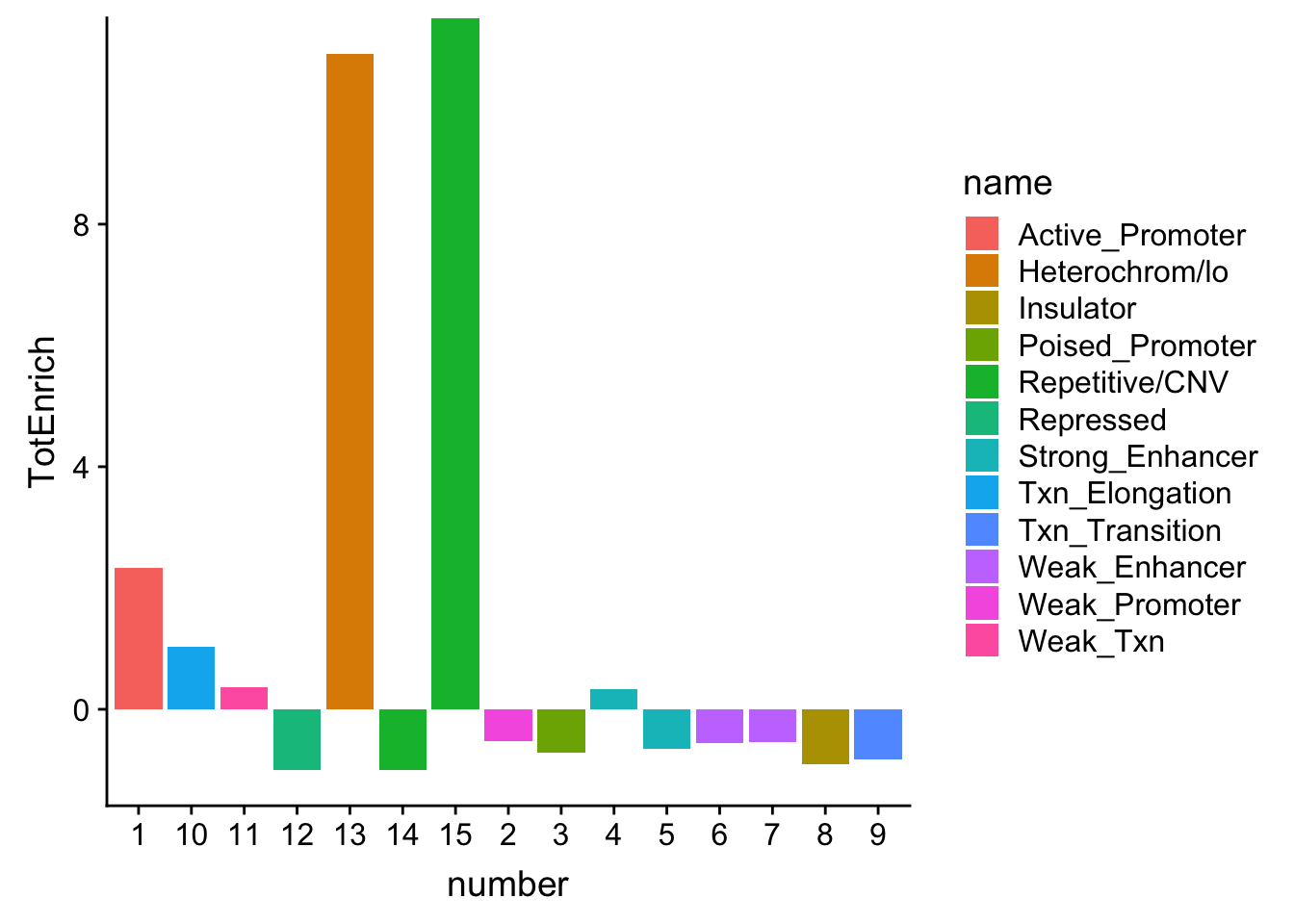
Expand here to see past versions of unnamed-chunk-49-2.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
Join together:
bothEnrich=NuclearOverlapHMM_enrichment %>% full_join(TotalOverlapHMM_enrichment, by=c("name", "number")) %>% select(number, name, NucEnrich,TotEnrich)
bothEnrich_melt=melt(bothEnrich, id.vars=c("number", "name"))ggplot(bothEnrich_melt, aes(x=number, by=variable, fill=name, y=value,col=variable)) + geom_bar(position = "dodge", stat = "identity",alpha=.5) + scale_color_manual(values=c("darkviolet", "deepskyblue3")) + labs(y="Enrichment from 1000 permutations", title="ChromHMM enrichment for \nTotal and Nuclear ApaQTLs",x="Region")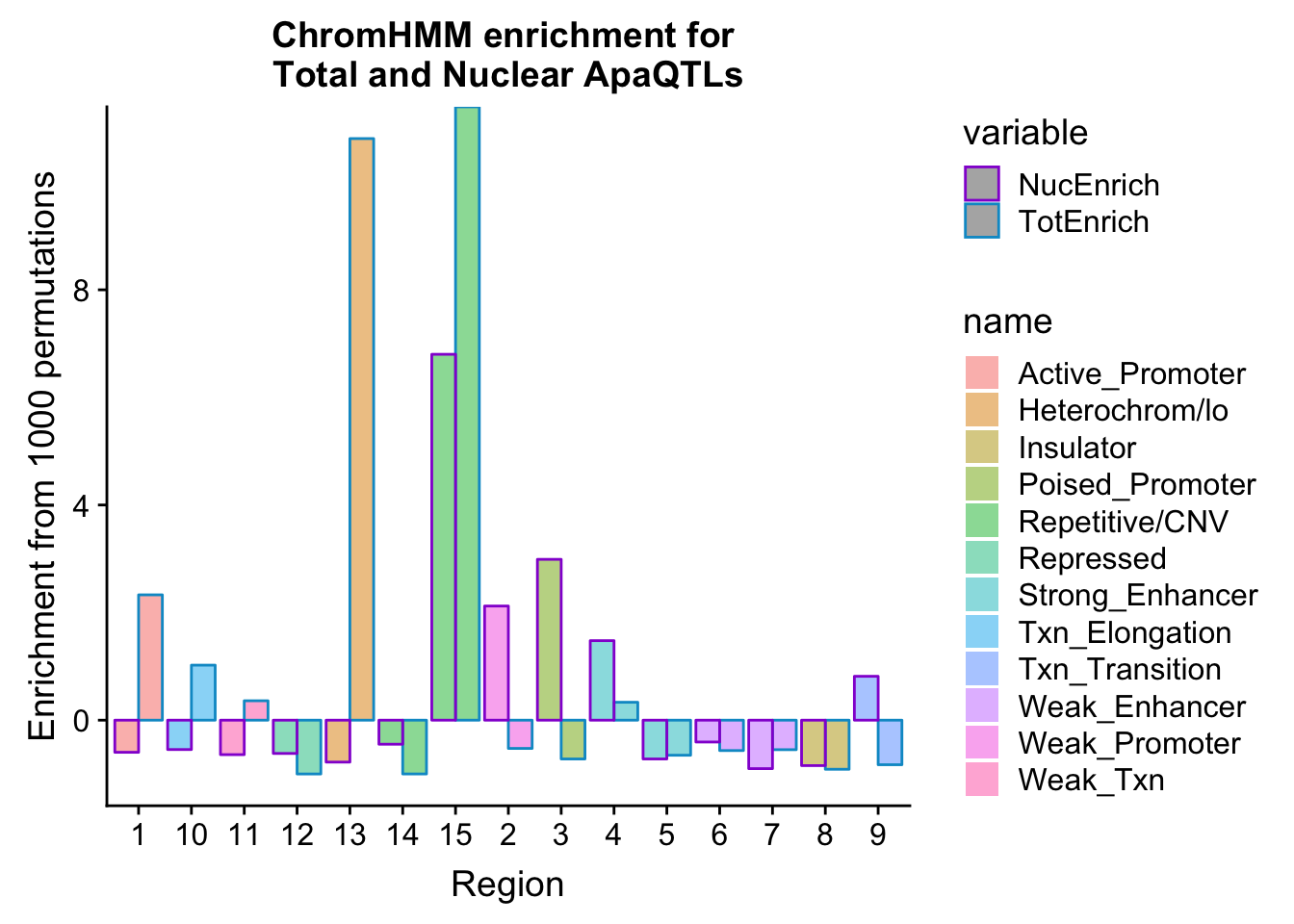
Expand here to see past versions of unnamed-chunk-51-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
Look only at the interesting ones by subsetting:
bothEnrich_melt_filt=bothEnrich_melt %>% filter(str_detect(name,"Active_Promoter|Txn_Elongation|Weak_Txn|Heterochrom/lo|Weak_Promoter|Poised_Promoter|Txn_Transition"))
ggplot(bothEnrich_melt_filt, aes(x=name, by=variable, fill=variable, y=value))+ geom_bar(position = "dodge", stat = "identity") + scale_fill_manual(values=c("deepskyblue3","darkviolet")) + theme(axis.text.x = element_text(angle = 90, hjust = 1)) + labs(y="Enrichment", x="Category", title="ChromHMM categroies \n with oppositte Enrichemtn patterns")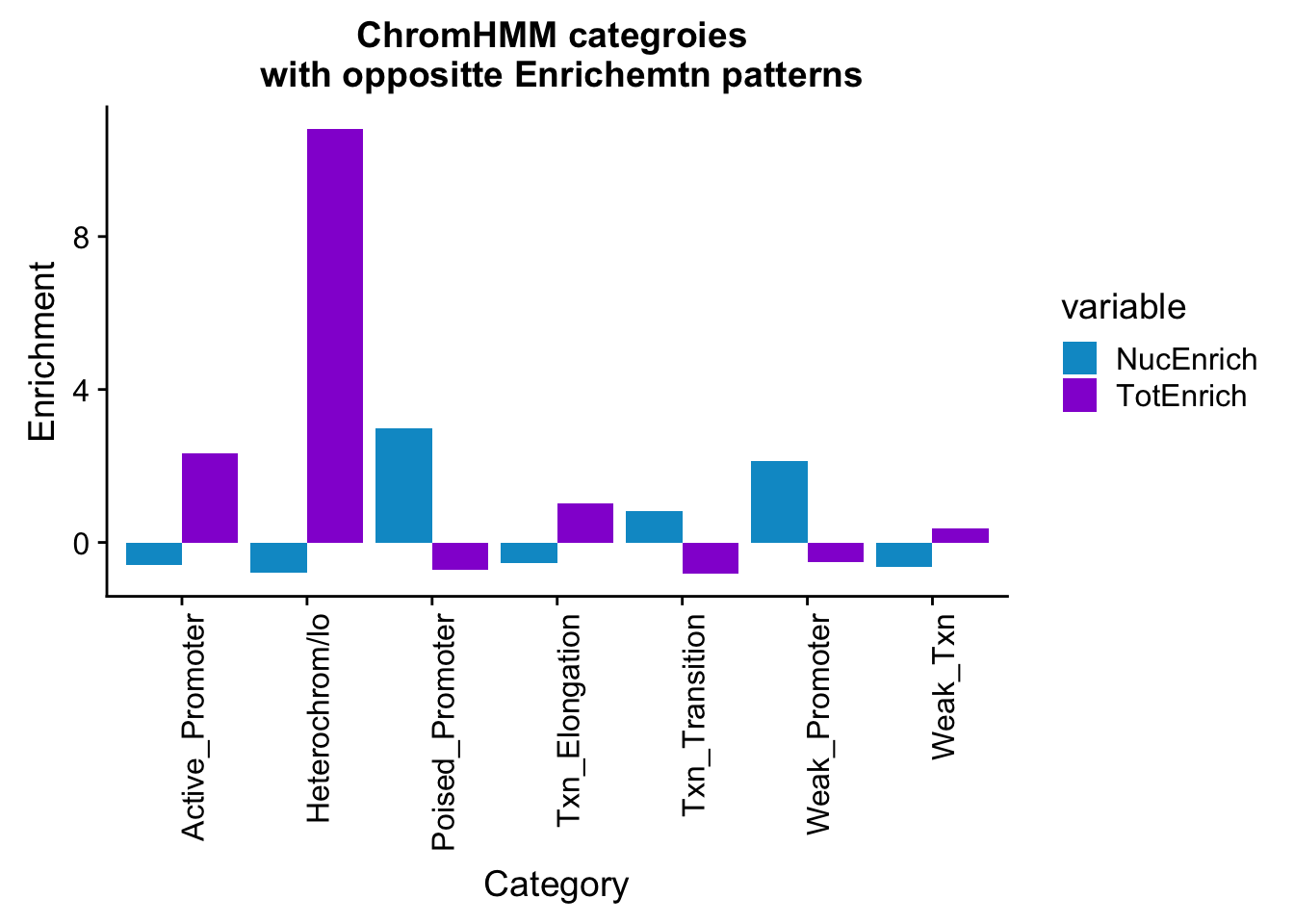
Expand here to see past versions of unnamed-chunk-52-1.png:
| Version | Author | Date |
|---|---|---|
| 83f1b14 | Briana Mittleman | 2018-11-08 |
The bimodal distributions may come from including both the significant and non significant genes in the test set. I need to remove all of the lines that come from a gene with a significant peak.
Remove significant genes
NucQTL_genes=read.table("../data/perm_QTL_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_transcript_permResBH.txt", stringsAsFactors = F, header=T) %>% mutate(sig=ifelse(-log10(bh)>=1, 1,0 )) %>% separate(pid, sep = ":", into=c("chr", "start", "end", "id")) %>% separate(id, sep = "_", into=c("gene", "strand", "peak")) %>% filter(sig==1) %>% select(gene) %>% distinct(gene)
#715 genes
#write this out as NucAPAGenes
write.table(NucQTL_genes, "../data/perm_QTL_trans/NucApaGenes.txt", row.names = F, col.names = F, quote=F)
TotQTL_genes=read.table("../data/perm_QTL_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_transcript_permResBH.txt", stringsAsFactors = F, header=T) %>% mutate(sig=ifelse(-log10(bh)>=1, 1,0 )) %>% separate(pid, sep = ":", into=c("chr", "start", "end", "id")) %>% separate(id, sep = "_", into=c("gene", "strand", "peak")) %>% filter(sig==1) %>% select(gene) %>% distinct(gene)
#106 genes
#write out as TotAPAGenes
write.table(TotQTL_genes, "../data/perm_QTL_trans/TotApaGenes.txt", row.names = F, col.names = F, quote=F)I need to find a way to get rid of these from the files I cam pulling from.
/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt
/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_NomRes.txtI can create an python script to do this. I will need to seperate the first column and and only write the line out if the gene is in the apaGenes files I just created.
filterSigGenes.py
#python
#genes with sig ApaQTL
TotGenes=open("/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/sig_genes/TotApaGenes.txt", "r")
NucGenes=open("/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/sig_genes/NucApaGenes.txt", "r")
#nom res (with all snps tested)
NucRes=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Nuclear_NomRes.txt", "r")
TotRes=open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/filtered_APApeaks_merged_allchrom_refseqGenes_pheno_Total_NomRes.txt", "r")
#output files:
Nuc_nonSig=open("/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/NucTestedSnps_nonSigGenes.txt", "w")
Tot_nonSig=open("/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/TotTestedSnps_nonSigGenes.txt", "w")
#convert genes to list
def file_to_list(file):
gene_list=[]
for ln in file:
gene=ln.strip()
gene_list.append(gene)
return(gene_list)
Tot_gene_list=file_to_list(TotGenes)
Nuc_gene_list=file_to_list(NucGenes)
#function that will take in the input, the list, and the output. I want to be able to run this function for total and nuclear
def filter(fin,fout, sigGenes):
for ln in fin:
gene=ln.split()[0].split(":")[3].split("_")[0]
if gene not in sigGenes:
fout.write(ln)
fout.close()
filter(NucRes,Nuc_nonSig,Nuc_gene_list)
filter(TotRes, Tot_nonSig, Tot_gene_list)
Call this in a bash script:
run_filterSigGenes.sh
#!/bin/bash
#SBATCH --job-name=run_filterSigGenes
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=run_filterSigGenes.out
#SBATCH --error=run_filterSigGenes.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python filterSigGenes.py
nuc_random880_chromHmm_noSig_set1.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_noSig_set1
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_noSig_set1.out
#SBATCH --error=nuc_random880_chromHmm_noSig_set1.err
#SBATCH --partition=bigmem2
#SBATCH --mem=100G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {1..250};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/NucTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/randomRes_Nuclear_880_noSig_${i}.txt
done
nuc_random880_chromHmm_noSig_set2.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_noSig_set2
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_noSig_set2.out
#SBATCH --error=nuc_random880_chromHmm_noSig_set2.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {251..500};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/NucTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/randomRes_Nuclear_880_noSig_${i}.txt
done
nuc_random880_chromHmm_noSig_set3.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_noSig_set3
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_noSig_set3.out
#SBATCH --error=nuc_random880_chromHmm_noSig_set3.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {501..750};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/NucTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/randomRes_Nuclear_880_noSig_${i}.txt
done
nuc_random880_chromHmm_noSig_set4.sh
#!/bin/bash
#SBATCH --job-name=nuc_random880_chromHmm_noSig_set4
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=nuc_random880_chromHmm_noSig_set4.out
#SBATCH --error=nuc_random880_chromHmm_noSig_set4.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {751..1000};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/NucTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/randomRes_Nuclear_880_noSig_${i}.txt
done
Same for total:
total_random118_chromHmm_noSig_set1.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_noSig_set1
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_noSig_set1.out
#SBATCH --error=total_random118_chromHmm_noSig_set1.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {1..250};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/TotTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/randomRes_Total_118_noSig_${i}.txt
done
total_random118_chromHmm_noSig_set2.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_noSig_set2
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_noSig_set2.out
#SBATCH --error=total_random118_chromHmm_noSig_set2.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {251..500};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/TotTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/randomRes_Total_118_noSig_${i}.txt
done
total_random118_chromHmm_noSig_set3.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_noSig_set3
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_noSig_set3.out
#SBATCH --error=total_random118_chromHmm_noSig_set3.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {501..750};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/TotTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/randomRes_Total_118_noSig_${i}.txt
done
total_random118_chromHmm_noSig_set4.sh
#!/bin/bash
#SBATCH --job-name=total_random118_chromHmm_noSig_set4
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=total_random118_chromHmm_noSig_set4.out
#SBATCH --error=total_random118_chromHmm_noSig_set4.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#test with 2 permutations then make it 1000
#choose random res
for i in {751..1000};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/TotTestedSnps_nonSigGenes.txt > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/randomRes_Total_118_noSig_${i}.txt
done
This may not be enough. I may need to change this so it only has uniq snp. I could be sampling the same snps over an over.
Make these files into snp bed files:
randomRes2SNPbed_noSig.py
def main(inFile, outFile):
fout=open(outFile, "w")
fin=open(inFile, "r")
for ln in fin:
pid, sid, dist, pval, slope = ln.split()
chrom, pos= sid.split(":")
name=sid
start= int(pos)-1
end=int(pos)
strand=pid.split(":")[3].split("_")[1]
pval=float(pval)
fout.write("%s\t%s\t%s\t%s\t%s\t%s\n"%(chrom, start, end, name, pval, strand))
fout.close()
if __name__ == "__main__":
import sys
fraction=sys.argv[1]
nsamp=sys.argv[2]
nsamp=int(nsamp)
iter=sys.argv[3]
inFile = "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s_noSig/randomRes_%s_%d_noSig_%s.txt"%(fraction,fraction, nsamp, iter)
outFile= "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s_noSig/snp_bed_noSig/randomRes_%s_%d_noSig_%s.bed"%(fraction,fraction, nsamp, iter)
main(inFile,outFile)
randomLines2Snp_noSig.sh
#!/bin/bash
#SBATCH --job-name=randomLines2Snp_noSig
#SBATCH --account=pi-gilad
#SBATCH --time=36:00:00
#SBATCH --output=randomLines2Snp_noSig.out
#SBATCH --error=randomLines2Snp_noSig.err
#SBATCH --partition=gilad
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
#make random
for i in {1..1000};
do
python randomRes2SNPbed_noSig.py Nuclear 880 ${i}
done
#make random
for i in {1..1000};
do
python randomRes2SNPbed_noSig.py Total 118 ${i}
done sortRandomSnps_noSig.sh
#!/bin/bash
#SBATCH --job-name=sortRandomSnps_noSig
#SBATCH --account=pi-yangili1
#SBATCH --time=10:00:00
#SBATCH --output=sortRandomSnps_noSig.out
#SBATCH --error=sortRandomSnps_noSig.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in $(ls /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/snp_bed_noSig/);
do
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/snp_bed_noSig/$i > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/snp_bed_sort_noSig/$i.sort.bed
done
for i in $(ls /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/snp_bed_noSig/);
do
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/snp_bed_noSig/$i > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/snp_bed_sort_noSig/$i.sort.bed
done
overlap_chromHMM_sepfiles_noSig.py
def main(inFile, outFile):
rand=pybedtools.BedTool(inFile)
hmm=pybedtools.BedTool("/project2/gilad/briana/genome_anotation_data/GM12878.chromHMM.sort.bed")
#map hmm to snps
Rand_overlapHMM=rand.map(hmm, c=4)
#save results
Rand_overlapHMM.saveas(outFile)
if __name__ == "__main__":
import sys
import pandas as pd
import pybedtools
fraction=sys.argv[1]
nsamp=sys.argv[2]
niter=sys.argv[3]
#which itteration we are on
inFile ="/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s_noSig/snp_bed_sort_noSig/randomRes_%s_%s_noSig_%s.bed.sort.bed"%(fraction,fraction, nsamp, niter)
outFile= "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s_noSig/chromHMM_overlap/randomres_overlapChromHMM_%s_%s_noSig_%s.txt"%(fraction,fraction,nsamp, niter)
main(inFile,outFile)overlap_chromHMM_sepfiles_noSig.sh
#!/bin/bash
#SBATCH --job-name=overlap_chromHMM_sepfiles_noSig
#SBATCH --account=pi-yangili1
#SBATCH --time=10:00:00
#SBATCH --output=overlap_chromHMM_sepfiles_noSig.out
#SBATCH --error=overlap_chromHMM_sepfiles_noSig.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
python overlap_chromHMM_sepfiles_noSig.py Nuclear 880 $i
done
for i in {1..1000};
do
python overlap_chromHMM_sepfiles_noSig.py Total 118 $i
donegroupRandomChromHMM_noSig.sh
#!/bin/bash
#SBATCH --job-name=groupRandomChromHMM_noSig
#SBATCH --account=pi-yangili1
#SBATCH --time=5:00:00
#SBATCH --output=groupRandomChromHMM.out
#SBATCH --error=groupRandomChromHMM.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
Rscript groupRandomByChromHMM.R -f /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/chromHMM_overlap/randomres_overlapChromHMM_Nuclear_880_noSig_${i}.txt -o /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_noSig/chromHMM_overlap_group/randomres_overlapChromHMM_Nuclear_880_noSig_${i}_grouped.txt
done
for i in {1..1000};
do
Rscript groupRandomByChromHMM.R -f /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/chromHMM_overlap/randomres_overlapChromHMM_Total_118_noSig_${i}.txt -o /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_noSig/chromHMM_overlap_group/randomres_overlapChromHMM_Total_118_noSig_${i}_grouped.txt
done
cut -d$' ' -f 1,2 randomres_overlapChromHMM_Nuclear_880_noSig_549_grouped.txt > Nuc_chromOverlap_noSig.txt
for i in {1..1000};
do
paste -d" " Nuc_chromOverlap_noSig.txt <(cut -d" " -f 3 randomres_overlapChromHMM_Nuclear_880_noSig_${i}_grouped.txt) > tmp
mv tmp Nuc_chromOverlap_noSig.txt
done
cut -d$' ' -f 1,2 randomres_overlapChromHMM_Total_118_noSig_53_grouped.txt > Tot_chromOverlap_noSig.txt
for i in {1..1000};
do
paste -d" " Tot_chromOverlap_noSig.txt <(cut -d" " -f 3 randomres_overlapChromHMM_Total_118_noSig_${i}_grouped.txt) > tmp
mv tmp Tot_chromOverlap_noSig.txt
doneThese files are not on my computer so I can work with them.
permutationResTotal_noSig=read.table("../data/ChromHmmOverlap/Tot_chromOverlap_noSig.txt", header=T, stringsAsFactors = F)
permutationResTotal_noSig[is.na(permutationResTotal_noSig)] <- 0
permutationResTotal_noSig= as_data_frame(permutationResTotal_noSig)
permutationResTotal_noSig_noName=permutationResTotal_noSig[,3:ncol(permutationResTotal_noSig)]
totRand_mean_noSig=rowMeans(permutationResTotal_noSig_noName)/1000
permutationResNuclear_noSig=read.table("../data/ChromHmmOverlap/Nuc_chromOverlap_noSig.txt",header = T,stringsAsFactors = F)
permutationResNuclear_noSig[is.na(permutationResNuclear_noSig)] <- 0
permutationResNuclear_noSig_noName=permutationResNuclear_noSig[,3:ncol(permutationResNuclear_noSig)]
nucRand_mean_noSig=rowMeans(permutationResNuclear_noSig_noName)/1000permutationResTotal_noSig_melt= melt(permutationResTotal_noSig, id.vars=c("number", "name"))ggplot(permutationResTotal_noSig_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + labs(x="N random Snps in category", title="Random permutation Total")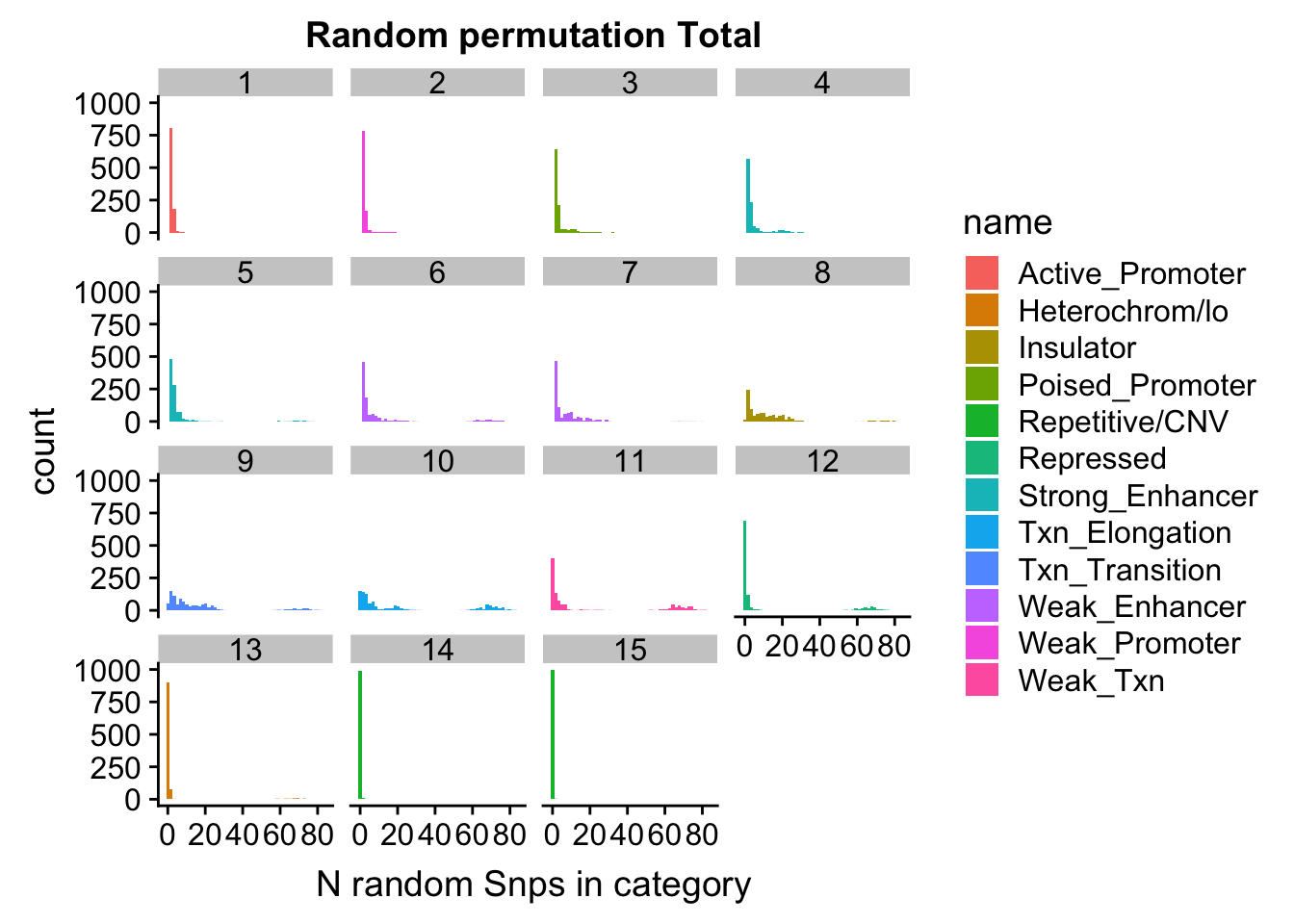
Expand here to see past versions of unnamed-chunk-74-1.png:
| Version | Author | Date |
|---|---|---|
| 086fbcc | Briana Mittleman | 2018-11-13 |
For nuclear:
permutationResNuclear_noSig_melt= melt(permutationResNuclear_noSig, id.vars=c("number", "name"))ggplot(permutationResNuclear_noSig_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + labs(x="N random Snps in category", title="Random permutation Nuclear")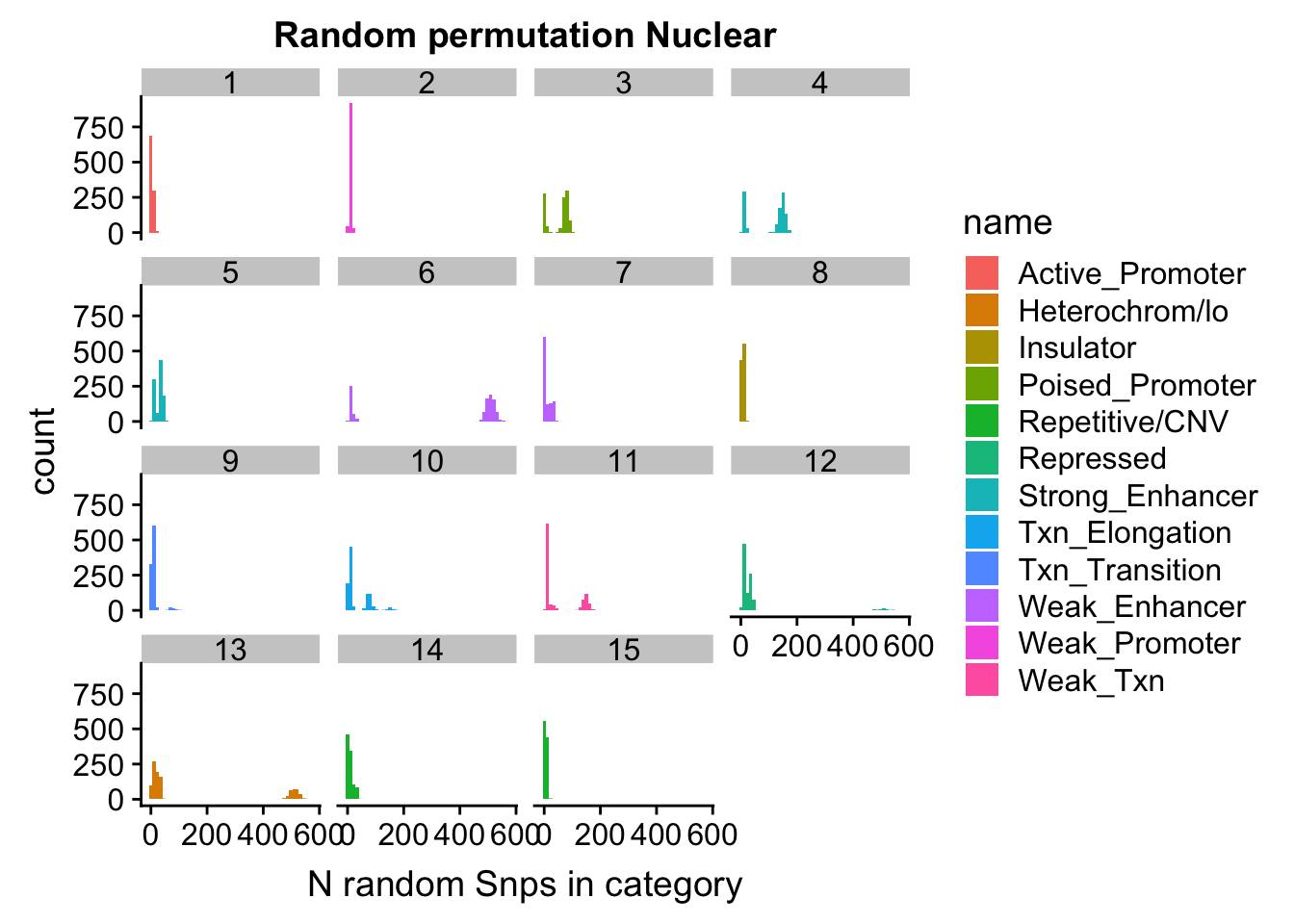
Expand here to see past versions of unnamed-chunk-76-1.png:
| Version | Author | Date |
|---|---|---|
| 086fbcc | Briana Mittleman | 2018-11-13 |
ggplot(permutationResNuclear_noSig_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + scale_y_log10()+ labs(x="random Snps in category", title="Random permutation Nuclear")Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 630 rows containing missing values (geom_bar).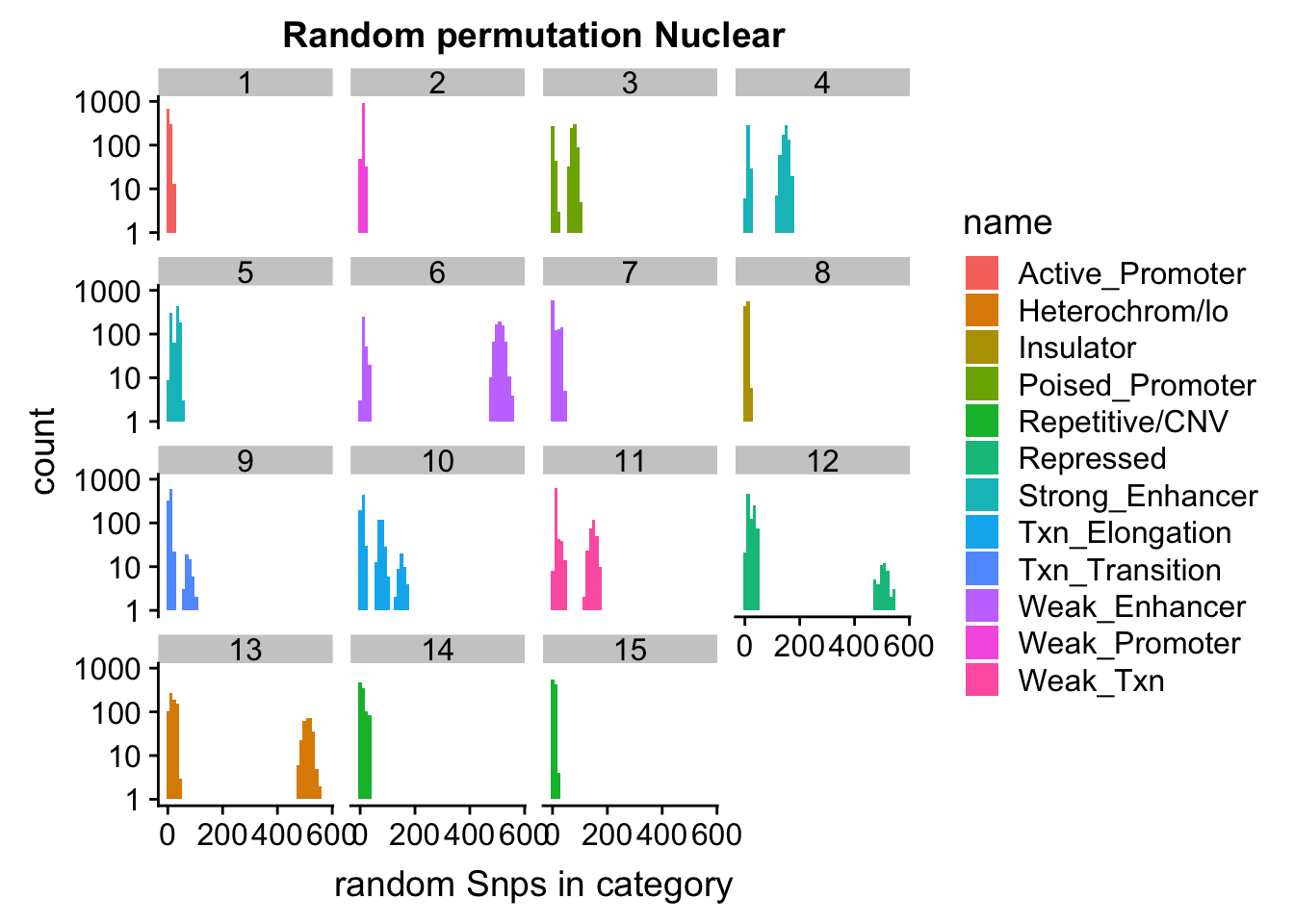
Expand here to see past versions of unnamed-chunk-77-1.png:
| Version | Author | Date |
|---|---|---|
| 086fbcc | Briana Mittleman | 2018-11-13 |
ggplot(permutationResTotal_noSig_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + scale_y_log10()+ labs(x="random Snps in category", title="Random permutation Total")Warning: Transformation introduced infinite values in continuous y-axisWarning: Removed 445 rows containing missing values (geom_bar).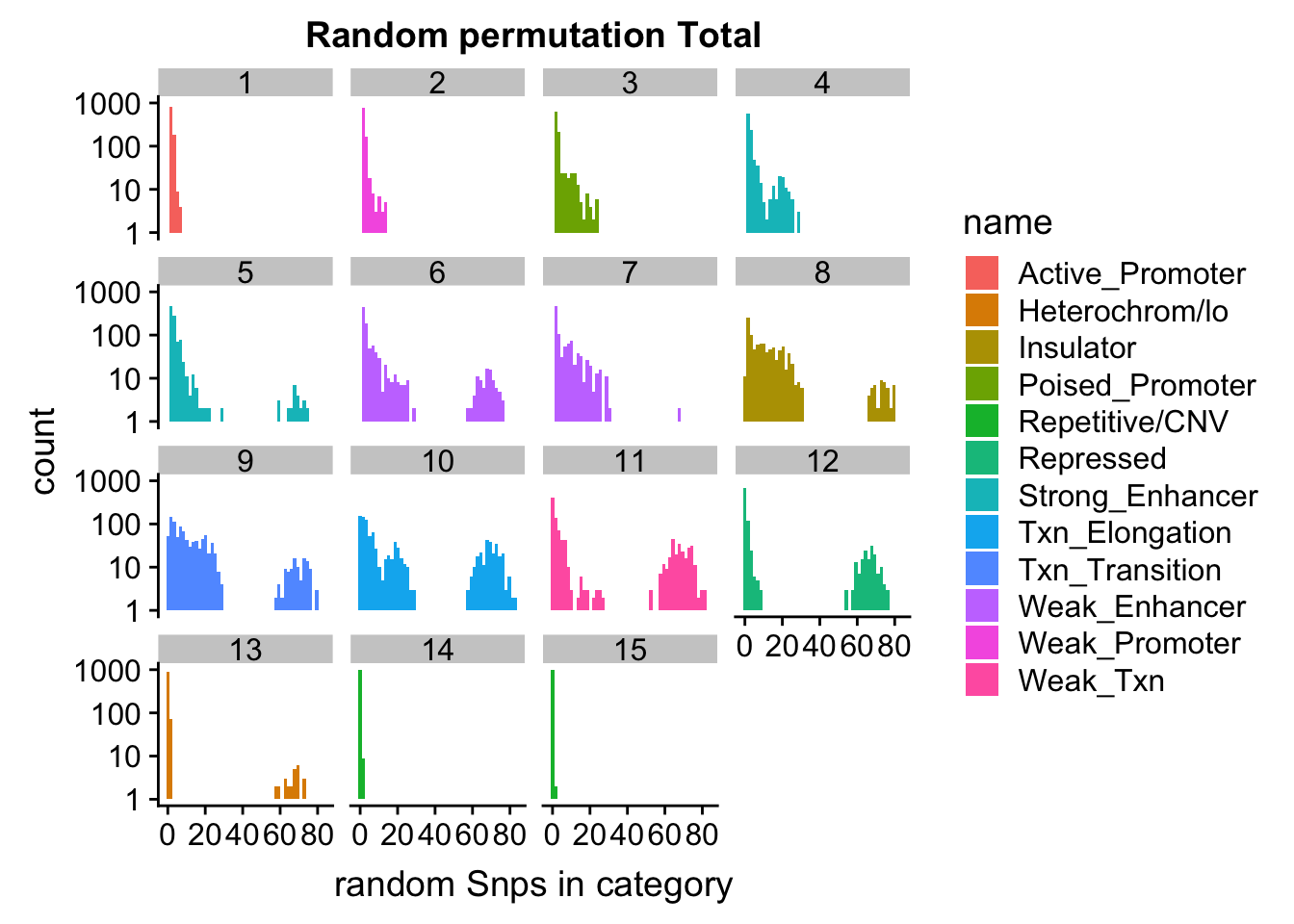
Expand here to see past versions of unnamed-chunk-77-2.png:
| Version | Author | Date |
|---|---|---|
| 086fbcc | Briana Mittleman | 2018-11-13 |
Permute only from unique snps tested
This didnt help. I need to choose from the unique snps rather than counting if they were tested multiple times. I will look for the snps tested and get rid of those called as QTLs.
Steps:
- get total and nuclear snps tested
I need to do this from the nominal results in /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/. The snp ID is in the second column
run_testedSnps_noSig.sh
#!/bin/bash
#SBATCH --job-name=run_testedSnps_noSig.
#SBATCH --account=pi-yangili1
#SBATCH --time=10:00:00
#SBATCH --output=run_testedSnps_noSig.out
#SBATCH --error=run_testedSnps_noSig.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
cut -f2 -d" " /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/NucTestedSnps_nonSigGenes.txt | sort | uniq > /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/uniq_tested_Nuclear.txt
cut -f2 -d" " /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/nomRes_nonsig/TotTestedSnps_nonSigGenes.txt | sort | uniq > /project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/uniq_tested_Total.txt- remove QTL snps
The significant QTL snps are:
/project2/gilad/briana/threeprimeseq/data/GWAS_overlap/ApaQTLsigSNPpos_Nuclear.txt /project2/gilad/briana/threeprimeseq/data/GWAS_overlap/ApaQTLsigSNPpos_Total.txt
I can use python to remove the snps from the full lists:
I can write it as a bed file to make the step easier.
testedSnps_noSig.py
total_qtl=open('/project2/gilad/briana/threeprimeseq/data/GWAS_overlap/ApaQTLsigSNPpos_Total.txt', "r")
nuclear_qtl=open('/project2/gilad/briana/threeprimeseq/data/GWAS_overlap/ApaQTLsigSNPpos_Total.txt', "r")
def file_to_list(file):
snp_list=[]
for ln in file:
snp=ln.strip()
snp_list.append(snp)
return(snp_list)
total_qtl_list= file_to_list(total_qtl)
nuclear_qtl_list= file_to_list(nuclear_qtl)
total_out=open("/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/uniqNonSigSnps/TotalUniqTestedSnp.bed","w")
for ln in open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/uniq_tested_Total.txt"):
snp=ln.strip()
if snp not in total_qtl_list:
chrom, pos =snp.split(":")
start=int(pos)-1
end=int(pos)
total_out.write("%s\t%d\t%d\t%s\n"%(chrom, start, end, snp))
total_out.close()
nuclear_out=open("/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/uniqNonSigSnps/NuclearUniqTestedSnp.bed","w")
for ln in open("/project2/gilad/briana/threeprimeseq/data/nominal_APAqtl_trans/uniq_tested_Nuclear.txt"):
snp=ln.strip()
if snp not in total_qtl_list:
chrom, pos =snp.split(":")
start=int(pos)-1
end=int(pos)
nuclear_out.write("%s\t%d\t%d\t%s\n"%(chrom, start, end, snp))
nuclear_out.close()
I will probablly need to run this in a bash script.
run_testedSnps2bed.sh
#!/bin/bash
#SBATCH --job-name=run_testedSnps2bed.sh
#SBATCH --account=pi-yangili1
#SBATCH --time=10:00:00
#SBATCH --output=run_testedSnps2bed.out
#SBATCH --error=run_testedSnps2bed.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
python testedSnps_noSig.py - select correct number of snps 1000 times (permutation) This file is not as big. I may be able to run all of the permutations at once:
random_UniqNoSig.sh
#!/bin/bash
#SBATCH --job-name=random_UniqNoSig
#SBATCH --account=pi-yangili1
#SBATCH --time=36:00:00
#SBATCH --output=random_UniqNoSig.out
#SBATCH --error=random_UniqNoSig.err
#SBATCH --partition=bigmem2
#SBATCH --mem=200G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
shuf -n 118 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/uniqNonSigSnps/TotalUniqTestedSnp.bed > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/snp_bed/randomRes_Total_118_UniqnoSig_${i}.bed
done
for i in {1..1000};
do
shuf -n 880 /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/uniqNonSigSnps/NuclearUniqTestedSnp.bed > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/snp_bed/randomRes_Nuclear_880_UniqnoSig_${i}.bed
done- sort bed files
sortRandomUniqSnps_noSig.sh
#!/bin/bash
#SBATCH --job-name=sortRandomUniqSnps_noSig
#SBATCH --account=pi-yangili1
#SBATCH --time=10:00:00
#SBATCH --output=sortRandomUniqSnps_noSig.out
#SBATCH --error=sortRandomUniqSnps_noSig.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in $(ls /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/snp_bed);
do
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/snp_bed/$i > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/snp_bed_sort_noSig/$i.sort.bed
done
for i in $(ls /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/snp_bed);
do
sort -k1,1 -k2,2n /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/snp_bed/$i > /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/snp_bed_sort_noSig/$i.sort.bed
done
- overlap each with chrom HMM
overlap_chromHMM_sepfiles_UniqnoSig.py
def main(inFile, outFile):
rand=pybedtools.BedTool(inFile)
hmm=pybedtools.BedTool("/project2/gilad/briana/genome_anotation_data/GM12878.chromHMM.sort.bed")
#map hmm to snps
Rand_overlapHMM=rand.map(hmm, c=4)
#save results
Rand_overlapHMM.saveas(outFile)
if __name__ == "__main__":
import sys
import pandas as pd
import pybedtools
fraction=sys.argv[1]
nsamp=sys.argv[2]
niter=sys.argv[3]
#which itteration we are on
inFile ="/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s_Uniq_noSig/snp_bed_sort_noSig/randomRes_%s_%s_UniqnoSig_%s.bed.sort.bed"%(fraction,fraction, nsamp, niter)
outFile= "/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/%s_Uniq_noSig/chromHMM_overlap/randomres_overlapChromHMM_%s_%s_UniqnoSig_%s.txt"%(fraction,fraction,nsamp, niter)
main(inFile,outFile)overlap_chromHMM_sepfiles_UniqnoSig.sh
#!/bin/bash
#SBATCH --job-name=overlap_chromHMM_sepfiles_UniqnoSig
#SBATCH --account=pi-yangili1
#SBATCH --time=10:00:00
#SBATCH --output=overlap_chromHMM_sepfiles_UniqnoSig.out
#SBATCH --error=overlap_chromHMM_sepfiles_UniqnoSig.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
python overlap_chromHMM_sepfiles_UniqnoSig.py Nuclear 880 $i
done
for i in {1..1000};
do
python overlap_chromHMM_sepfiles_UniqnoSig.py Total 118 $i
done- group the chrom HMM calls
I need to modify groupRandomByChromHMM_uniq.R
#!/bin/rscripts
# usage: groupRandomByChromHMM_uniq.R -f infile -o outfile
#this file will take any of the itterations and output a file with chrom hmm number, name, numberof snps
library(optparse)
library(dplyr)
library(tidyr)
library(ggplot2)
library(readr)
option_list = list(
make_option(c("-f", "--file"), action="store", default=NA, type='character',
help="input coverage file"),
make_option(c("-o", "--output"), action="store", default=NA, type='character',
help="output file")
)
opt_parser <- OptionParser(option_list=option_list)
opt <- parse_args(opt_parser)
#interrupt execution if no file is supplied
if (is.null(opt$file)){
print_help(opt_parser)
stop("Need input file", call.=FALSE)
}
if (is.null(opt$output)){
print_help(opt_parser)
stop("Need output file", call.=FALSE)
}
randomSNPS=read.table(opt$file, col.names=c("chrom", "start", "end", "sid", "number"),stringsAsFactors = F, na.strings = "NA")
hmm_names=read.table("/project2/gilad/briana/genome_anotation_data/chromHMM_regions.txt", col.names = c("number", "name"),stringsAsFactors=F)
randomSNPS$number=as.character(randomSNPS$number)
hmm_names$number=as.character(hmm_names$number)
randomSNPS_names= randomSNPS %>% left_join(hmm_names, by="number")
#split the name of the file to get the iteration number
fileSplit=strsplit(opt$file, "/")[[1]][10]
iter.txt=strsplit(fileSplit, "_")[[1]][5]
iter=substr(iter.txt, 1, nchar(iter.txt)-4)
randomSNPS_names_grouped=randomSNPS_names %>% group_by(number) %>% summarise(!!iter:=n()) %>% replace_na(list(name="No_annotation")) %>% dplyr::select(number, !!iter)
hmm_names$number=as.character(hmm_names$number)
final=hmm_names %>% left_join(randomSNPS_names_grouped,by="number")
write.table(final,opt$output,quote=FALSE, col.names = T, row.names = F)groupRandomChromHMM_UniqnoSig.sh
#!/bin/bash
#SBATCH --job-name=groupRandomChromHMM_UniqnoSig
#SBATCH --account=pi-yangili1
#SBATCH --time=5:00:00
#SBATCH --output=groupRandomChromHMM_UniqnoSig.out
#SBATCH --error=groupRandomChromHMM_UniqnoSig.err
#SBATCH --partition=broadwl
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in {1..1000};
do
Rscript groupRandomByChromHMM_uniq.R -f /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/chromHMM_overlap/randomres_overlapChromHMM_Nuclear_880_UniqnoSig_${i}.txt -o /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/chromHMM_overlap_group/randomres_overlapChromHMM_Nuclear_880_UniqnoSig_${i}_grouped.txt
done
for i in {1..1000};
do
Rscript groupRandomByChromHMM_uniq.R -f /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/chromHMM_overlap/randomres_overlapChromHMM_Total_118_UniqnoSig_${i}.txt -o /project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/chromHMM_overlap_group/randomres_overlapChromHMM_Total_118_UniqnoSig_${i}_grouped.txt
done
- make the final matrix
#/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Nuclear_Uniq_noSig/chromHMM_overlap_group/
cut -d$' ' -f 1,2 randomres_overlapChromHMM_Nuclear_880_UniqnoSig_549_grouped.txt > Nuc_chromOverlap_UniqnoSig.txt
for i in {1..1000};
do
paste -d" " Nuc_chromOverlap_UniqnoSig.txt <(cut -d" " -f 3 randomres_overlapChromHMM_Nuclear_880_UniqnoSig_${i}_grouped.txt) > tmp
mv tmp Nuc_chromOverlap_UniqnoSig.txt
done
#/project2/gilad/briana/threeprimeseq/data/random_QTLsnps/Total_Uniq_noSig/chromHMM_overlap_group/
cut -d$' ' -f 1,2 randomres_overlapChromHMM_Total_118_UniqnoSig_53_grouped.txt > Tot_chromOverlap_UniqnoSig.txt
for i in {1..1000};
do
paste -d" " Tot_chromOverlap_UniqnoSig.txt <(cut -d" " -f 3 randomres_overlapChromHMM_Total_118_UniqnoSig_${i}_grouped.txt) > tmp
mv tmp Tot_chromOverlap_UniqnoSig.txt
donepermutationResTotal_UniqnoSig=read.table("../data/ChromHmmOverlap/Tot_chromOverlap_UniqnoSig.txt", header=T, stringsAsFactors = F)
permutationResTotal_UniqnoSig[is.na(permutationResTotal_UniqnoSig)] <- 0
permutationResTotal_UniqnoSig= as_data_frame(permutationResTotal_UniqnoSig)
permutationResTotal_UniqnoSig_noName=permutationResTotal_UniqnoSig[,3:ncol(permutationResTotal_UniqnoSig)]
totRand_mean_UniqnoSig=rowMeans(permutationResTotal_UniqnoSig_noName)/1000
permutationResNuclear_UniqnoSig=read.table("../data/ChromHmmOverlap/Nuc_chromOverlap_UniqnoSig.txt",header = T,stringsAsFactors = F)
permutationResNuclear_UniqnoSig[is.na(permutationResNuclear_UniqnoSig)] <- 0
permutationResNuclear_UniqnoSig_noName=permutationResNuclear_UniqnoSig[,3:ncol(permutationResNuclear_UniqnoSig)]
nucRand_mean_UniqnoSig=rowMeans(permutationResNuclear_UniqnoSig_noName)/1000permutationResTotal_UniqnoSig_melt= melt(permutationResTotal_UniqnoSig, id.vars=c("number", "name"))ggplot(permutationResTotal_UniqnoSig_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + labs(x="N random Snps in category", title="Random permutation Total")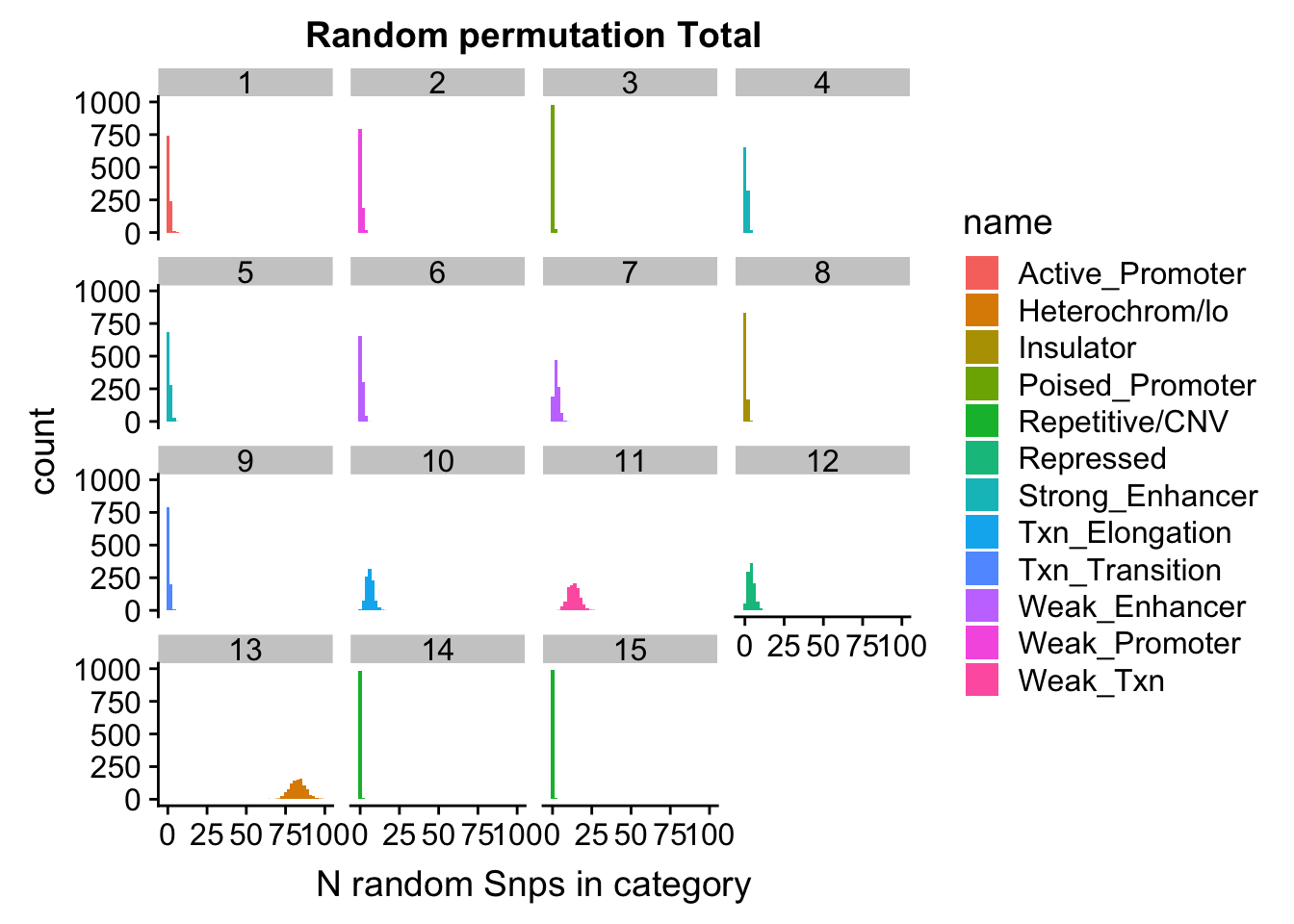
permutationResNuclear_UniqnoSig_melt= melt(permutationResNuclear_UniqnoSig, id.vars=c("number", "name"))ggplot(permutationResNuclear_UniqnoSig_melt, aes(x=value,fill=name)) + geom_histogram(bins=50) + facet_wrap(~number) + labs(x="N random Snps in category", title="Random permutation Total")
This is the model I will move forward with.
Plot the enrichment:
Look at enrichment by using the average
TotalPermMean_uniq=permutationResTotal_UniqnoSig_melt %>% group_by(number) %>% summarise(TotRandPerm=mean(value), TotRandPermSD=sd(value))
TotalPermMean_uniq$number=as.character(TotalPermMean_uniq$number)
NuclearPermMean_uniq=permutationResNuclear_UniqnoSig_melt %>% group_by(number) %>% summarise(NucRandPerm=mean(value),NucRandPermSD=sd(value))
NuclearPermMean_uniq$number=as.character(NuclearPermMean_uniq$number)Melt SNP values by name and number to get data in same format. I already did this above.
TotalOverlapHMM_names_melt=melt(TotalOverlapHMM_names, id.vars=c("number", "name"))%>% filter(variable=="sid") %>% group_by(number) %>% summarise(TotalQTL=n())
TotalOverlapHMM_names_melt$number=as.character(TotalOverlapHMM_names_melt$number)
NuclearOverlapHMM_names_melt=melt(NuclearOverlapHMM_names, id.vars=c("number", "name")) %>% filter(variable=="sid") %>% group_by(number) %>% summarise(NucQTL=n())
NuclearOverlapHMM_names_melt$number=as.character(NuclearOverlapHMM_names_melt$number)chromHmm$number=as.character(chromHmm$number)
TotalOverlapHMM_Uniq_enrichment= TotalOverlapHMM_names_melt %>% full_join(TotalPermMean_uniq, by="number") %>% replace_na(list(TotalQTL=.00001)) %>% full_join(chromHmm, by="number")
TotalOverlapHMM_Uniq_enrichment$TotalQTL=as.double(TotalOverlapHMM_Uniq_enrichment$TotalQTL)
TotalOverlapHMM_Uniq_enrichment = TotalOverlapHMM_Uniq_enrichment %>% mutate(TotEnrich=(TotalQTL-TotRandPerm)/TotRandPermSD)
NuclearOverlapHMM_Uniq_enrichment=NuclearOverlapHMM_names_melt %>% full_join(NuclearPermMean_uniq, by="number")%>% full_join(chromHmm, by="number")
NuclearOverlapHMM_Uniq_enrichment$NucQTL=as.double(NuclearOverlapHMM_Uniq_enrichment$NucQTL)
NuclearOverlapHMM_Uniq_enrichment=NuclearOverlapHMM_Uniq_enrichment %>%mutate(NucEnrich=(NucQTL-NucRandPerm)/NucRandPermSD)ggplot(NuclearOverlapHMM_Uniq_enrichment, aes(y=NucEnrich, x=number, fill=name)) + geom_bar(stat="identity") + labs(title="Nuclear ApaQTL enrichment Z score", y="Enrichment Z score")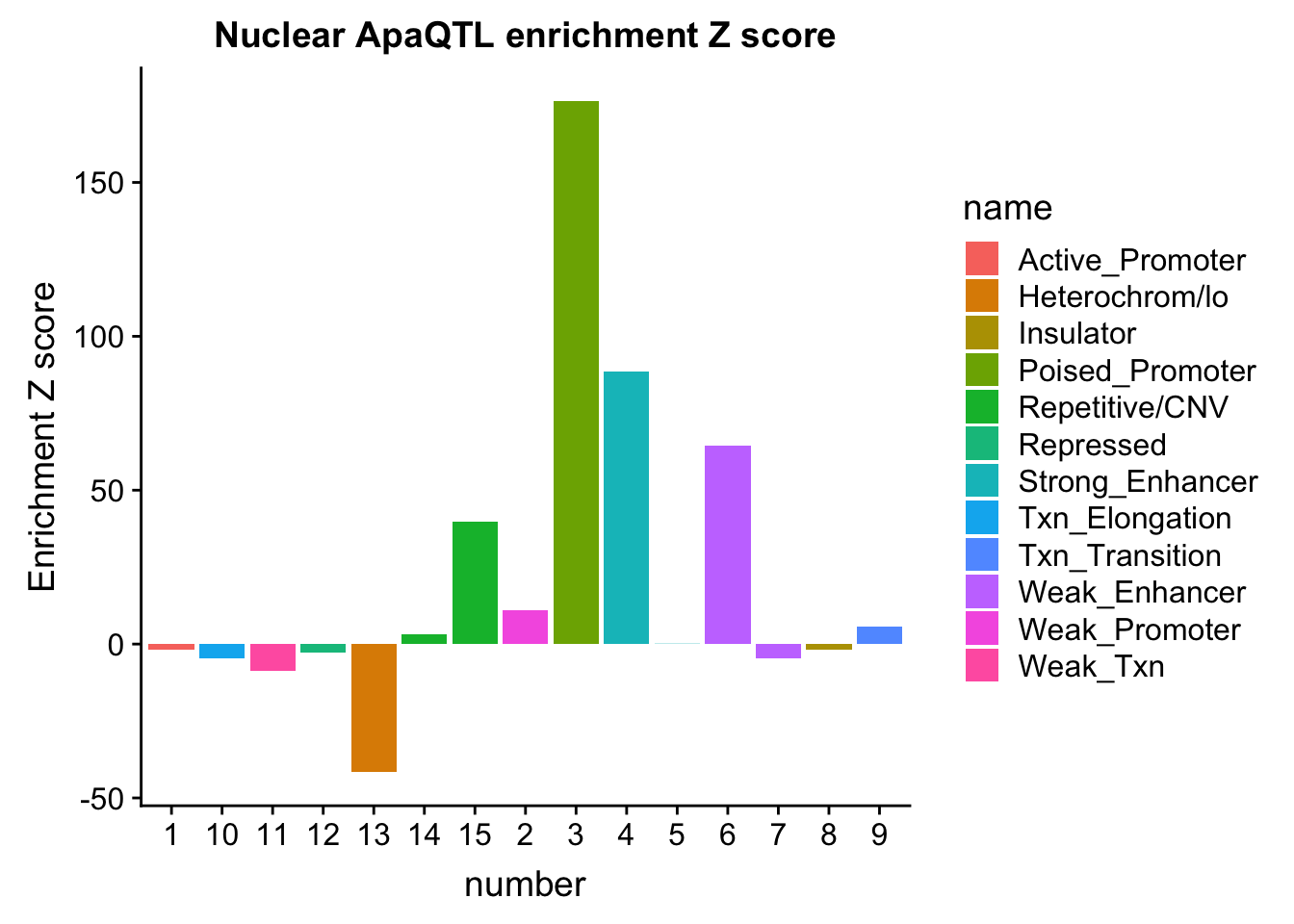
ggplot(TotalOverlapHMM_Uniq_enrichment, aes(y=TotEnrich, x=number, fill=name)) + geom_bar(stat="identity")+ labs(title="Total ApaQTL enrichment Z score", y="Enrichment Z score")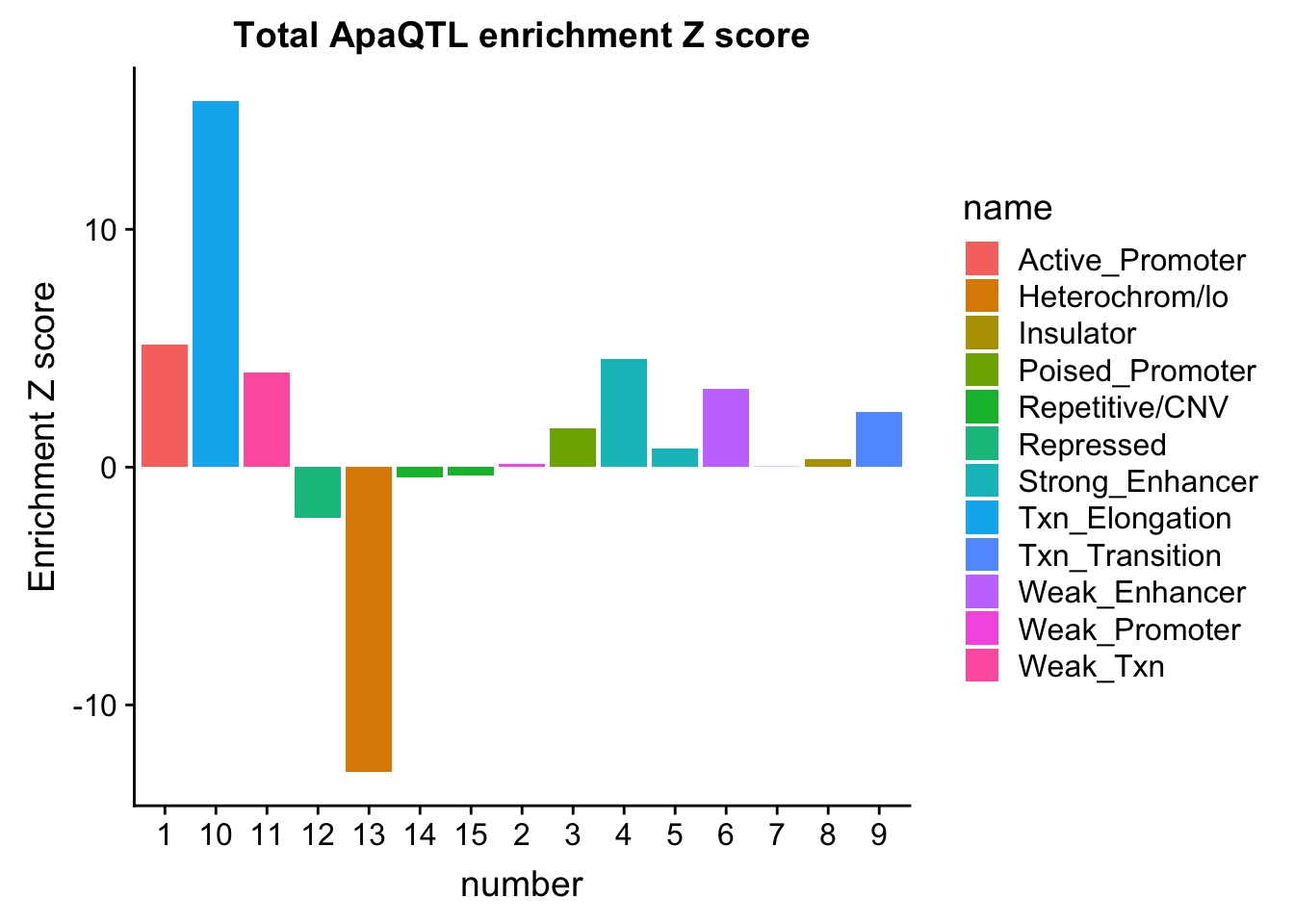
bothEnrich_uniq=NuclearOverlapHMM_Uniq_enrichment %>% full_join(TotalOverlapHMM_Uniq_enrichment, by=c("name", "number")) %>% select(number, name, NucEnrich,TotEnrich)
bothEnrich_uniqmelt=melt(bothEnrich_uniq, id.vars=c("number", "name"))ggplot(bothEnrich_uniqmelt, aes(x=number, by=variable, fill=variable, y=value,col=name)) + geom_bar(position = "dodge", stat = "identity",alpha=.5) + scale_fill_manual(values=c("darkviolet", "deepskyblue3")) + labs(y="Enrichment from 1000 permutations", title="ChromHMM enrichment for \nTotal and Nuclear ApaQTLs",x="Region")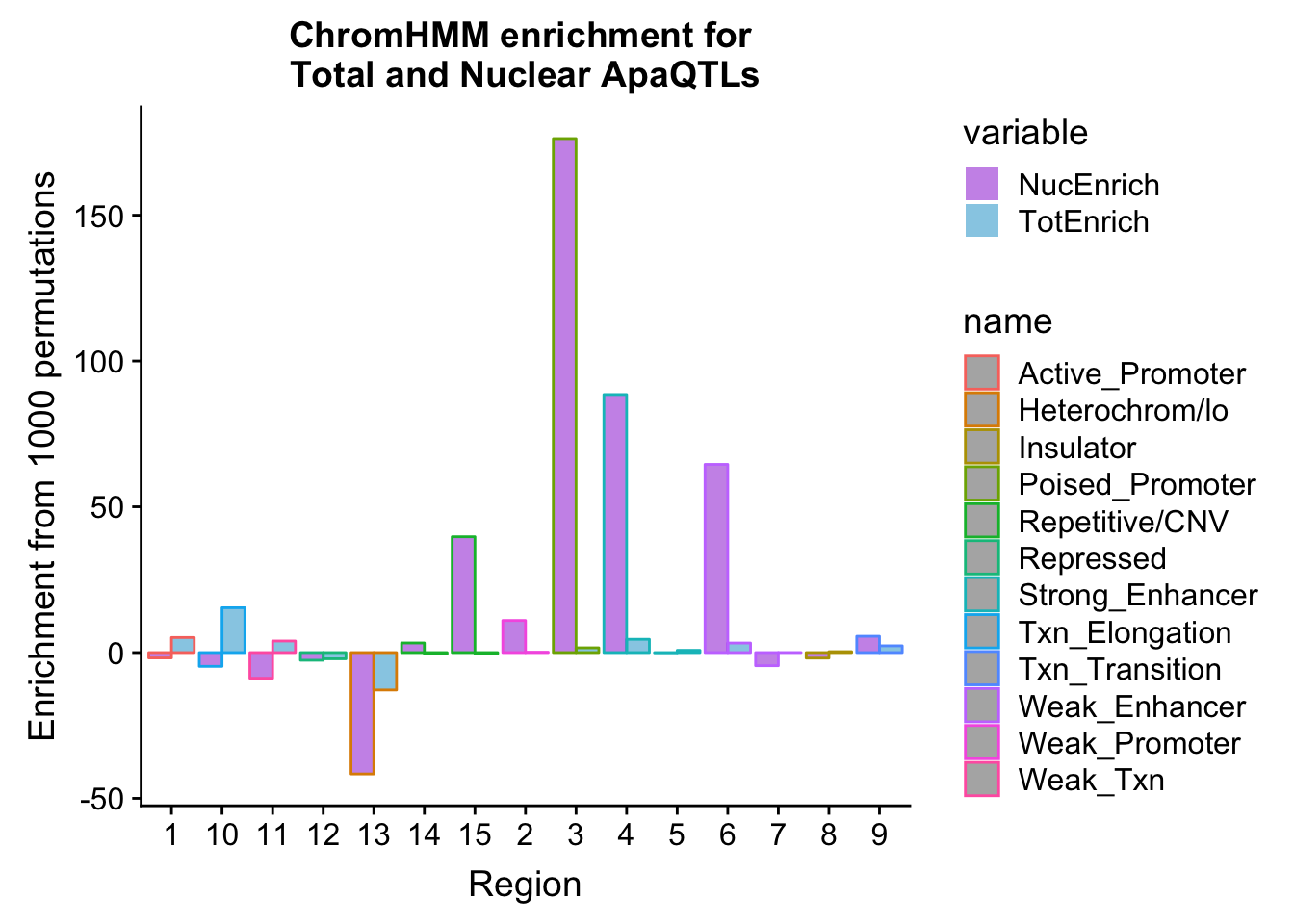
Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] bindrcpp_0.2.2 cowplot_0.9.3 ggpubr_0.1.8
[4] magrittr_1.5 data.table_1.11.8 VennDiagram_1.6.20
[7] futile.logger_1.4.3 forcats_0.3.0 stringr_1.3.1
[10] dplyr_0.7.6 purrr_0.2.5 readr_1.1.1
[13] tidyr_0.8.1 tibble_1.4.2 ggplot2_3.0.0
[16] tidyverse_1.2.1 reshape2_1.4.3 workflowr_1.1.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.4 haven_1.1.2 lattice_0.20-35
[4] colorspace_1.3-2 htmltools_0.3.6 yaml_2.2.0
[7] rlang_0.2.2 R.oo_1.22.0 pillar_1.3.0
[10] glue_1.3.0 withr_2.1.2 R.utils_2.7.0
[13] RColorBrewer_1.1-2 lambda.r_1.2.3 modelr_0.1.2
[16] readxl_1.1.0 bindr_0.1.1 plyr_1.8.4
[19] munsell_0.5.0 gtable_0.2.0 cellranger_1.1.0
[22] rvest_0.3.2 R.methodsS3_1.7.1 evaluate_0.11
[25] labeling_0.3 knitr_1.20 broom_0.5.0
[28] Rcpp_0.12.19 formatR_1.5 backports_1.1.2
[31] scales_1.0.0 jsonlite_1.5 hms_0.4.2
[34] digest_0.6.17 stringi_1.2.4 rprojroot_1.3-2
[37] cli_1.0.1 tools_3.5.1 lazyeval_0.2.1
[40] futile.options_1.0.1 crayon_1.3.4 whisker_0.3-2
[43] pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4
[46] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[49] rstudioapi_0.8 R6_2.3.0 nlme_3.1-137
[52] git2r_0.23.0 compiler_3.5.1
This reproducible R Markdown analysis was created with workflowr 1.1.1