Fraction Differences with Processed data
Briana Mittleman
1/22/2019
Last updated: 2019-01-25
workflowr checks: (Click a bullet for more information)-
✔ R Markdown file: up-to-date
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
-
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 78f24b9
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: .Rhistory Ignored: .Rproj.user/ Ignored: data/.DS_Store Ignored: output/.DS_Store Untracked files: Untracked: KalistoAbundance18486.txt Untracked: analysis/DirectionapaQTL.Rmd Untracked: analysis/EvaleQTLs.Rmd Untracked: analysis/YL_QTL_test.Rmd Untracked: analysis/ncbiRefSeq_sm.sort.mRNA.bed Untracked: analysis/snake.config.notes.Rmd Untracked: analysis/verifyBAM.Rmd Untracked: code/PeaksToCoverPerReads.py Untracked: code/strober_pc_pve_heatmap_func.R Untracked: data/18486.genecov.txt Untracked: data/APApeaksYL.total.inbrain.bed Untracked: data/ChromHmmOverlap/ Untracked: data/GM12878.chromHMM.bed Untracked: data/GM12878.chromHMM.txt Untracked: data/LianoglouLCL/ Untracked: data/LocusZoom/ Untracked: data/NuclearApaQTLs.txt Untracked: data/PeakCounts/ Untracked: data/PeakCounts_noMP_5perc/ Untracked: data/PeakUsage/ Untracked: data/PeakUsage_noMP/ Untracked: data/PeaksUsed/ Untracked: data/PeaksUsed_noMP_5percCov/ Untracked: data/RNAkalisto/ Untracked: data/TotalApaQTLs.txt Untracked: data/Totalpeaks_filtered_clean.bed Untracked: data/UnderstandPeaksQC/ Untracked: data/YL-SP-18486-T-combined-genecov.txt Untracked: data/YL-SP-18486-T_S9_R1_001-genecov.txt Untracked: data/YL_QTL_test/ Untracked: data/apaExamp/ Untracked: data/apaQTL_examp_noMP/ Untracked: data/bedgraph_peaks/ Untracked: data/bin200.5.T.nuccov.bed Untracked: data/bin200.Anuccov.bed Untracked: data/bin200.nuccov.bed Untracked: data/clean_peaks/ Untracked: data/comb_map_stats.csv Untracked: data/comb_map_stats.xlsx Untracked: data/comb_map_stats_39ind.csv Untracked: data/combined_reads_mapped_three_prime_seq.csv Untracked: data/diff_iso_proc/ Untracked: data/diff_iso_trans/ Untracked: data/ensemble_to_genename.txt Untracked: data/example_gene_peakQuant/ Untracked: data/explainProtVar/ Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.bed Untracked: data/filtered_APApeaks_merged_allchrom_refseqTrans.closest2End.noties.bed Untracked: data/first50lines_closest.txt Untracked: data/gencov.test.csv Untracked: data/gencov.test.txt Untracked: data/gencov_zero.test.csv Untracked: data/gencov_zero.test.txt Untracked: data/gene_cov/ Untracked: data/joined Untracked: data/leafcutter/ Untracked: data/merged_combined_YL-SP-threeprimeseq.bg Untracked: data/molPheno_noMP/ Untracked: data/mol_overlap/ Untracked: data/mol_pheno/ Untracked: data/nom_QTL/ Untracked: data/nom_QTL_opp/ Untracked: data/nom_QTL_trans/ Untracked: data/nuc6up/ Untracked: data/nuc_10up/ Untracked: data/other_qtls/ Untracked: data/pQTL_otherphen/ Untracked: data/peakPerRefSeqGene/ Untracked: data/perm_QTL/ Untracked: data/perm_QTL_opp/ Untracked: data/perm_QTL_trans/ Untracked: data/perm_QTL_trans_filt/ Untracked: data/perm_QTL_trans_noMP_5percov/ Untracked: data/protAndAPAlmRes.Rda Untracked: data/reads_mapped_three_prime_seq.csv Untracked: data/smash.cov.results.bed Untracked: data/smash.cov.results.csv Untracked: data/smash.cov.results.txt Untracked: data/smash_testregion/ Untracked: data/ssFC200.cov.bed Untracked: data/temp.file1 Untracked: data/temp.file2 Untracked: data/temp.gencov.test.txt Untracked: data/temp.gencov_zero.test.txt Untracked: data/threePrimeSeqMetaData.csv Untracked: output/picard/ Untracked: output/plots/ Untracked: output/qual.fig2.pdf Unstaged changes: Modified: analysis/28ind.peak.explore.Rmd Modified: analysis/CompareLianoglouData.Rmd Modified: analysis/apaQTLoverlapGWAS.Rmd Modified: analysis/cleanupdtseq.internalpriming.Rmd Modified: analysis/coloc_apaQTLs_protQTLs.Rmd Modified: analysis/dif.iso.usage.leafcutter.Rmd Modified: analysis/diff_iso_pipeline.Rmd Modified: analysis/explainpQTLs.Rmd Modified: analysis/explore.filters.Rmd Modified: analysis/flash2mash.Rmd Modified: analysis/mispriming_approach.Rmd Modified: analysis/overlapMolQTL.Rmd Modified: analysis/overlapMolQTL.opposite.Rmd Modified: analysis/overlap_qtls.Rmd Modified: analysis/peakOverlap_oppstrand.Rmd Modified: analysis/peakQCPPlots.Rmd Modified: analysis/pheno.leaf.comb.Rmd Modified: analysis/swarmPlots_QTLs.Rmd Modified: analysis/test.max2.Rmd Modified: analysis/understandPeaks.Rmd Modified: code/Snakefile
Expand here to see past versions:
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 78f24b9 | Briana Mittleman | 2019-01-25 | look at mid range, draw conclusion |
| html | 3698aba | Briana Mittleman | 2019-01-24 | Build site. |
| Rmd | 19ec099 | Briana Mittleman | 2019-01-24 | look at mid range examples |
| html | d86a41c | Briana Mittleman | 2019-01-24 | Build site. |
| Rmd | 82c1571 | Briana Mittleman | 2019-01-24 | look at top 15 red |
| html | aefc330 | Briana Mittleman | 2019-01-22 | Build site. |
| Rmd | fd184be | Briana Mittleman | 2019-01-22 | add code for leafcutter on processed |
library(workflowr)This is workflowr version 1.1.1
Run ?workflowr for help getting startedlibrary(tidyverse)── Attaching packages ────────────────────────────────────────────────────────────── tidyverse 1.2.1 ──✔ ggplot2 3.0.0 ✔ purrr 0.2.5
✔ tibble 1.4.2 ✔ dplyr 0.7.6
✔ tidyr 0.8.1 ✔ stringr 1.3.1
✔ readr 1.1.1 ✔ forcats 0.3.0── Conflicts ───────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()library(reshape2)
Attaching package: 'reshape2'The following object is masked from 'package:tidyr':
smithslibrary(cowplot)
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsaveIn this analysis https://brimittleman.github.io/threeprimeseq/PeakToGeneAssignment.html I ran an initial run of the leafcutter tool for differences between fractions. I will use the same pipeline here for the processed data.
This starts with running feature counts with all of the peaks. I will use peaks passing the filter in either the total or nucelar fraction. These peaks are in /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR.refseqTrans.closest2end.sm.fixed_5percCov.bed and were created using the filternamePeaks5percCov.py script. I need to make this into an SAF file for FC.
This file has chr, start, end, peakNu, cov, strand, transcript:gene, distance. For the SAF file I want GeneID, Chr, start, end, strand. The GeneID is peak#:chr:start:end:strand:gene
bed2saf_bothFrac_Processed.py
from misc_helper import *
fout = open("/project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR.refseqTrans.closest2end.sm.fixed_5percCov.SAF",'w')
fout.write("GeneID\tChr\tStart\tEnd\tStrand\n")
for ln in open("/project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR.refseqTrans.closest2end.sm.fixed_5percCov.bed"):
chrom, start, end, peakNum, cov, strand, trans, dist = ln.split()
gene=trans.split(":")[1]
ID = "peak%s:%s:%s:%s:%s:%s"%(peakNum,chrom,start, end,strand,gene)
fout.write("%s\t%s\t%s\t%s\t%s\n"%(ID, chrom, start, end, strand))
fout.close()
bothFrac_processed_FC.sh
#!/bin/bash
#SBATCH --job-name=bothFrac_processed_FC
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=bothFrac_processed_FCc.out
#SBATCH --error=bothFrac_processed_FC.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
featureCounts -O -a /project2/gilad/briana/threeprimeseq/data/mergedPeaks_noMP_filtered/Filtered_APApeaks_merged_allchrom_noMP.sort.named.noCHR.refseqTrans.closest2end.sm.fixed_5percCov.SAF -F SAF -o /project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed.fc /project2/gilad/briana/threeprimeseq/data/bam_NoMP_sort/*sort.bam -s 2Fix headers:
fix_head_fc_procBothFrac.py
#python
infile= open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed.fc", "r")
fout = open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed_fixed.fc",'w')
for line, i in enumerate(infile):
if line == 1:
i_list=i.split()
libraries = i_list[:6]
print(libraries)
for sample in i_list[6:]:
full = sample.split("/")[7]
samp= full.split("-")[2:4]
lim="_"
samp_st=lim.join(samp)
libraries.append(samp_st)
first_line= "\t".join(libraries)
fout.write(first_line + '\n')
else :
fout.write(i)
fout.close()fc2leafphen_processed.py
inFile= open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed_fixed.fc", "r")
outFile= open("/project2/gilad/briana/threeprimeseq/data/pheno_DiffIso_processed/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed_forLC.fc", "w")
for num, ln in enumerate(inFile):
if num == 1:
lines=ln.split()[6:]
outFile.write(" ".join(lines)+'\n')
if num > 1:
ID=ln.split()[0]
peak=ID.split(":")[0]
chrom=ID.split(":")[1]
start=ID.split(":")[2]
start=int(start)
end=ID.split(":")[3]
end=int(end)
strand=ID.split(":")[4]
gene=ID.split(":")[5]
new_ID="chr%s:%d:%d:%s"%(chrom, start, end, gene)
pheno=ln.split()[6:]
pheno.insert(0, new_ID)
outFile.write(" ".join(pheno)+'\n')
outFile.close() subset_diffisopheno_processed.py
def main(inFile, outFile, target):
ifile=open(inFile, "r")
ofile=open(outFile, "w")
target=int(target)
for num, ln in enumerate(ifile):
if num == 0:
ofile.write(ln)
else:
ID=ln.split()[0]
chrom=ID.split(":")[0][3:]
print(chrom)
chrom=int(chrom)
if chrom == target:
ofile.write(ln)
if __name__ == "__main__":
import sys
target = sys.argv[1]
inFile = "/project2/gilad/briana/threeprimeseq/data/pheno_DiffIso_processed/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed_forLC.fc"
outFile = "/project2/gilad/briana/threeprimeseq/data/pheno_DiffIso_processed/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed_forLC_%s.txt"%(target)
main(inFile, outFile, target)Run this with: run_subset_diffisopheno_processed.sh
#!/bin/bash
#SBATCH --job-name=run_subset_diffisopheno_processed
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_subset_diffisopheno_processed.out
#SBATCH --error=run_subset_diffisopheno_processed.err
#SBATCH --partition=broadwl
#SBATCH --mem=12G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
python subset_diffisopheno_processed.py $i
doneMake new sample list. I could use the old one but I want to have this pipeline work when I add individuals.
makeLCSampleList_processed.py
outfile=open("/project2/gilad/briana/threeprimeseq/data/pheno_DiffIso_processed/sample_groups.txt", "w")
infile=open("/project2/gilad/briana/threeprimeseq/data/filtPeakOppstrand_cov_processed_bothFrac/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed.fc", "r")
for line, i in enumerate(infile):
if line == 1:
i_list=i.split()
libraries=[]
for sample in i_list[6:]:
full = sample.split("/")[7]
samp= full.split("-")[2:4]
lim="_"
samp_st=lim.join(samp)
libraries.append(samp_st)
for l in libraries:
if l[-1] == "T":
outfile.write("%s\tTotal\n"%(l))
else:
outfile.write("%s\tNuclear\n"%(l))
else:
next
outfile.close()run_leafcutter_ds_bychrom_processed.sh
#!/bin/bash
#SBATCH --job-name=run_leafcutter_ds_bychrom_processed
#SBATCH --account=pi-yangili1
#SBATCH --time=24:00:00
#SBATCH --output=run_leafcutter_ds_bychrom_processed.out
#SBATCH --error=run_leafcutter_ds_bychrom_processed.err
#SBATCH --partition=bigmem2
#SBATCH --mem=50G
#SBATCH --mail-type=END
module load R
for i in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22
do
Rscript /project2/gilad/briana/davidaknowles-leafcutter-c3d9474/scripts/leafcutter_ds.R --num_threads 4 /project2/gilad/briana/threeprimeseq/data/pheno_DiffIso_processed/filtered_APApeaks_merged_allchrom_refseqGenes.Transcript_sm_quant_processed_forLC_${i}.txt /project2/gilad/briana/threeprimeseq/data/pheno_DiffIso_processed/sample_groups.txt -o /project2/gilad/briana/threeprimeseq/data/diff_iso_processed/TN_diff_isoform_chr${i}.txt
doneCat all of the signficance files and bring them to my computer to look at here.
diffIso=read.table("../data/diff_iso_proc/TN_diff_isoform_allChrom_clusterSig.txt", header = T,col.names = c("status", "loglr", "df", "p", "cluster", "p.adjust"),stringsAsFactors = F,sep="\t") %>% filter(status == "Success")
diffIso$p.adjust=as.numeric(as.character(diffIso$p.adjust))
qqplot(-log10(runif(nrow(diffIso))), -log10(diffIso$p.adjust),ylab="-log10 Total Adjusted Leafcutter pvalue", xlab="-log 10 Uniform expectation", main="Leafcutter differencial isoform analysis between fractions")
abline(0,1)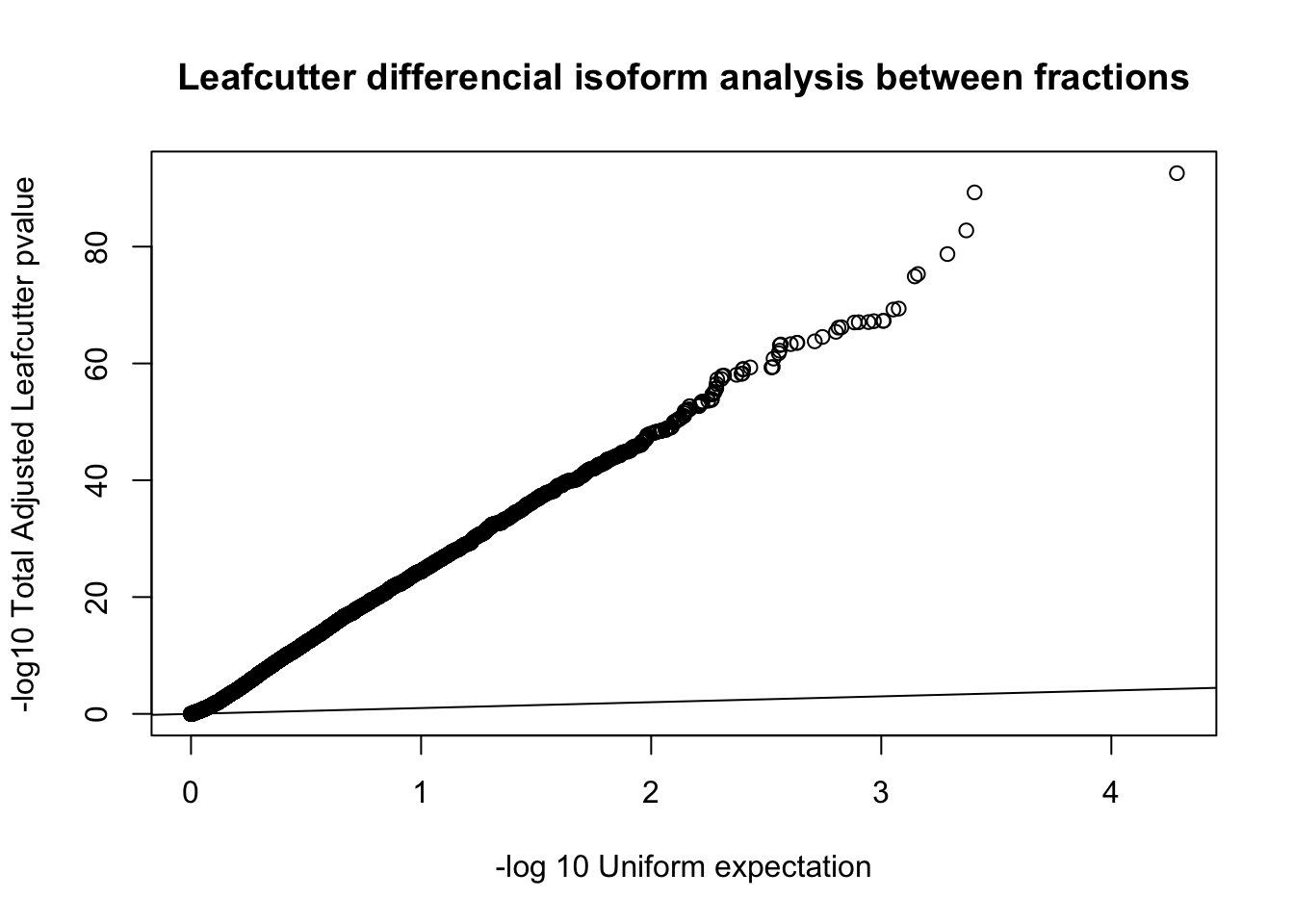
Expand here to see past versions of unnamed-chunk-10-1.png:
| Version | Author | Date |
|---|---|---|
| d86a41c | Briana Mittleman | 2019-01-24 |
This is based on successfull results for 9,532 genes with multiple PAS and enough coverage in enough individuals.
Next, I will look at the effect sizes.
awk '{if(NR>1)print}' /project2/gilad/briana/threeprimeseq/data/diff_iso_processed/TN_diff_isoform_chr*.txt_effect_sizes.txt > /project2/gilad/briana/threeprimeseq/data/diff_iso_processed/TN_diff_isoform_AllChrom.txt_effect_sizes.txteffectsize=read.table("../data/diff_iso_proc/TN_diff_isoform_AllChrom.txt_effect_sizes.txt", stringsAsFactors = F, col.names=c('intron', 'logef' ,'Nuclear', 'Total','deltapsi'))effectsize$logef=as.numeric(as.character(effectsize$logef))Warning: NAs introduced by coercionplot(sort(effectsize$logef),main="Leafcutter effect Sizes", ylab="Effect size", xlab="Peak Index")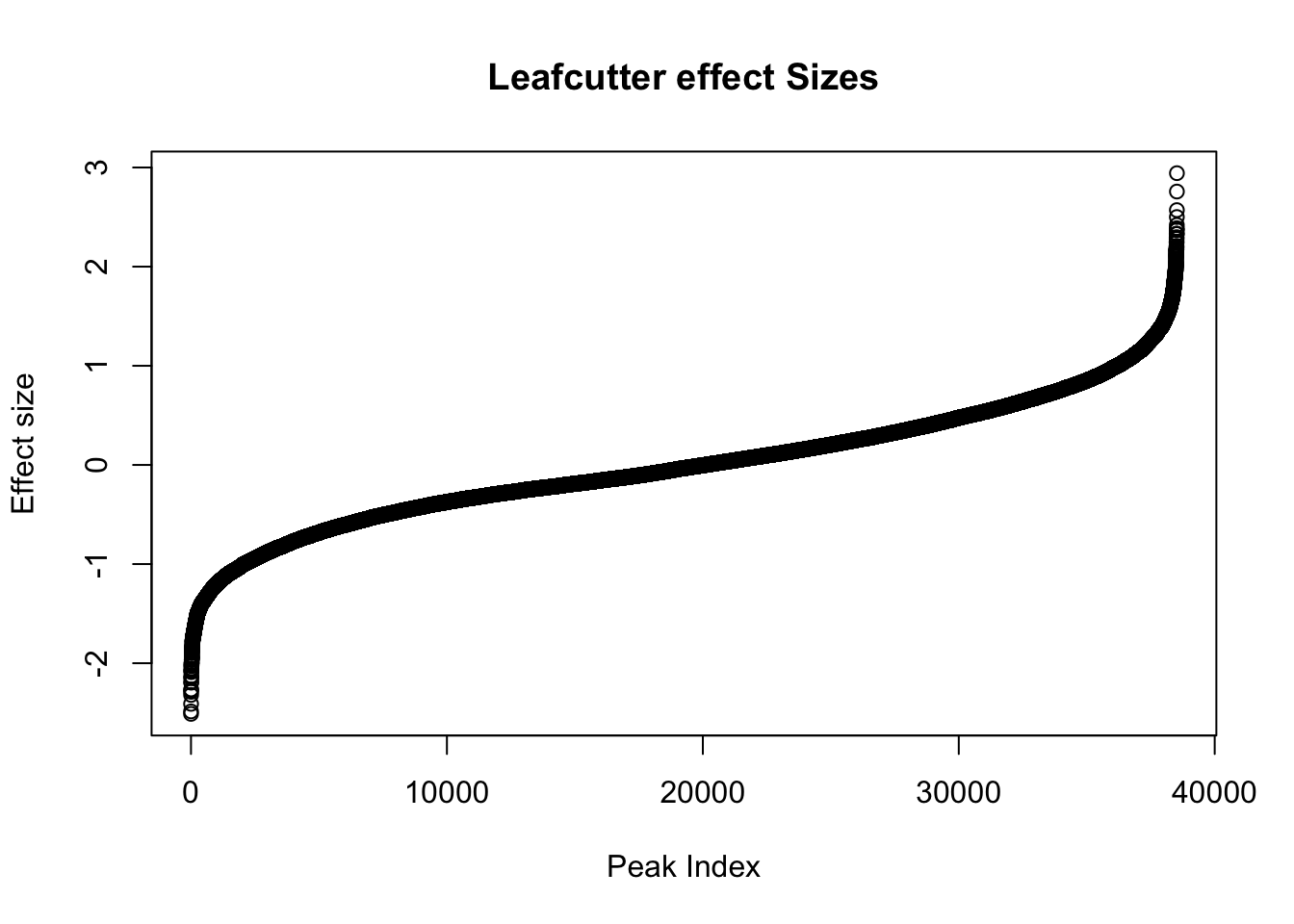
Expand here to see past versions of unnamed-chunk-13-1.png:
| Version | Author | Date |
|---|---|---|
| d86a41c | Briana Mittleman | 2019-01-24 |
effectsize$deltapsi=as.numeric(as.character(effectsize$deltapsi))Warning: NAs introduced by coercionplot(sort(effectsize$deltapsi),main="Leafcutter delta PSI", ylab="Delta PSI", xlab="Peak Index")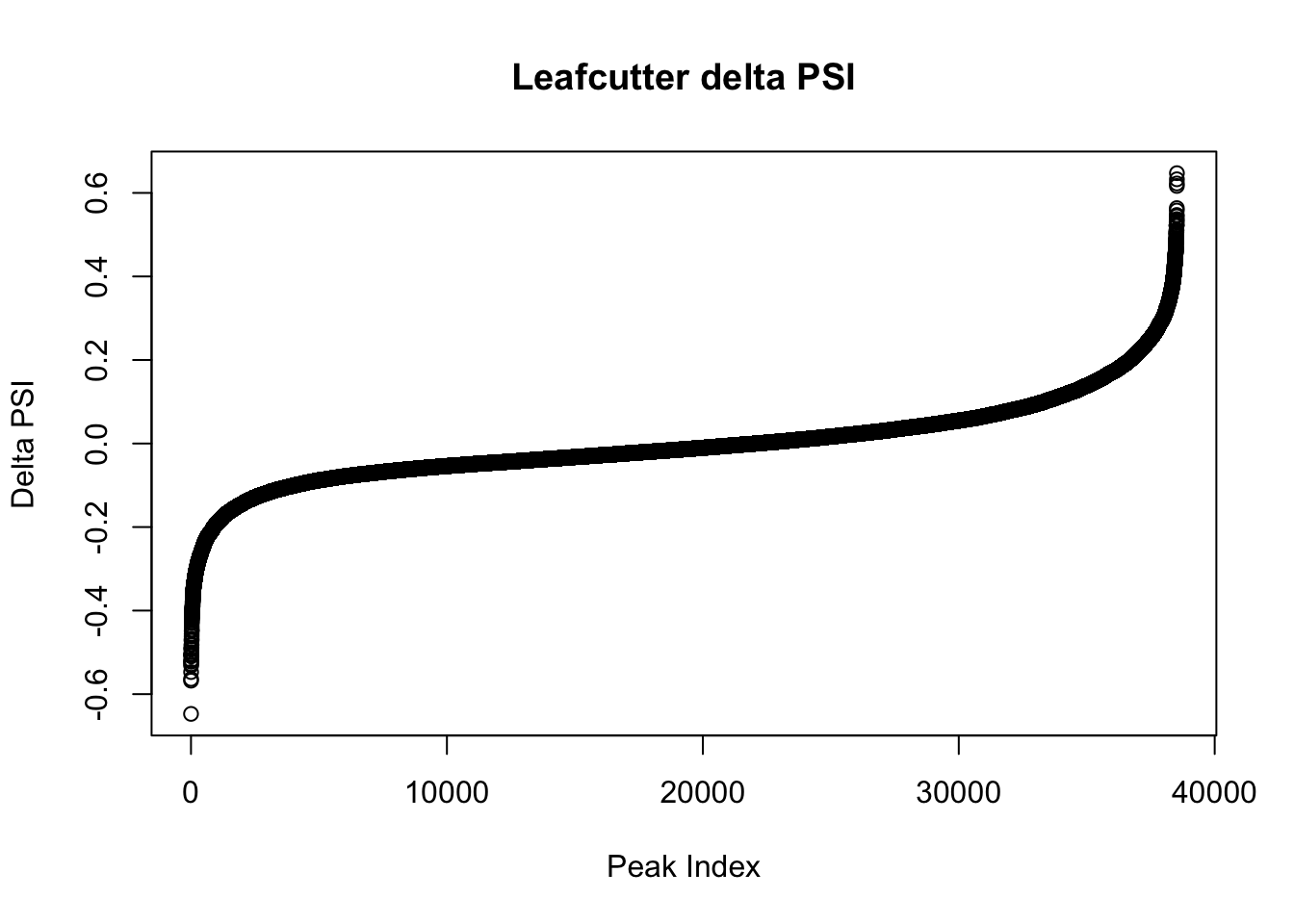
Expand here to see past versions of unnamed-chunk-14-1.png:
| Version | Author | Date |
|---|---|---|
| d86a41c | Briana Mittleman | 2019-01-24 |
I want to spot check some of these in IGV. First I will focus on the |PSI| >.4
filterHighPSI=effectsize %>% filter(abs(deltapsi)>.4) %>% arrange(deltapsi)
head(filterHighPSI) intron logef Nuclear
1 chr2:108464199:108464261:RGPD4 -1.563069 0.786325014667375
2 chr11:71409928:71409980:FAM86C1 -1.750522 0.736879355169336
3 chr9:19443338:19443405:SLC24A2 -1.373452 0.87662816529528
4 chr7:151725930:151726019:GALNTL5 -1.229096 0.766827065986407
5 chr17:49248730:49248817:NME2 -1.185121 0.763505415306066
6 chr7:100970341:100970385:IFT22 -1.588218 0.668693582230009
Total deltapsi
1 0.139047439399753 -0.6472776
2 0.169115629694331 -0.5677637
3 0.313023025601665 -0.5636051
4 0.219649286101815 -0.5471778
5 0.231787036297737 -0.5317184
6 0.140656353549046 -0.5280372Too look at this most effectively I will merge the total and nuclear clean bam files:
mergeBamFiles_byfrac_noMP.sh
#!/bin/bash
#SBATCH --job-name=mergeBamFiles_byfrac_noMP.sh
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=mergeBamFiles_byfrac_noMP.out
#SBATCH --error=mergeBamFiles_byfrac_noMP.err
#SBATCH --partition=bigmem2
#SBATCH --mem=100G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
samtools merge /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllTotalSamples.MergedBamFiles.noMP.bam /project2/gilad/briana/threeprimeseq/data/bam_NoMP_sort/*T*.bam
samtools merge /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllNuclearSamples.MergedBamFiles.noMP.bam /project2/gilad/briana/threeprimeseq/data/bam_NoMP_sort/*N*.bamSortIndex_mergeBamFiles_byfrac_noMP.sh
#!/bin/bash
#SBATCH --job-name=SortIndex_mergeBamFiles_byfrac_noMP.sh
#SBATCH --account=pi-yangili1
#SBATCH --time=8:00:00
#SBATCH --output=SortIndex_mergeBamFiles_byfrac_noMP.out
#SBATCH --error=SortIndex_mergeBamFiles_byfrac_noMP.err
#SBATCH --partition=bigmem2
#SBATCH --mem=100G
#SBATCH --mail-type=END
module load Anaconda3
source activate three-prime-env
samtools sort /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllTotalSamples.MergedBamFiles.noMP.bam > /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllTotalSamples.MergedBamFiles.noMP.sort.bam
samtools index /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllTotalSamples.MergedBamFiles.noMP.sort.bam
samtools sort /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllNuclearSamples.MergedBamFiles.noMP.bam > /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllNuclearSamples.MergedBamFiles.noMP.sort.bam
samtools index /project2/gilad/briana/threeprimeseq/data/mergedBams_NoMP/AllNuclearSamples.MergedBamFiles.noMP.sort.bamTake a look at some of the top hits in IGV:
slice(filterHighPSI,1:15) intron logef Nuclear
1 chr2:108464199:108464261:RGPD4 -1.563069 0.786325014667375
2 chr11:71409928:71409980:FAM86C1 -1.750522 0.736879355169336
3 chr9:19443338:19443405:SLC24A2 -1.373452 0.87662816529528
4 chr7:151725930:151726019:GALNTL5 -1.229096 0.766827065986407
5 chr17:49248730:49248817:NME2 -1.185121 0.763505415306066
6 chr7:100970341:100970385:IFT22 -1.588218 0.668693582230009
7 chr3:142422173:142422217:PCOLCE2 -1.160742 0.747193256154251
8 chr12:121337613:121337664:HNF1A -1.439657 0.922672953632586
9 chr8:134586015:134586097:WISP1 -1.828958 0.861221198355898
10 chr15:76234769:76234851:FBXO22 -1.494914 0.702484460762617
11 chr13:65049589:65049665:PCDH9 -1.585817 0.677762517045467
12 chr20:30651036:30651094:CCM2L -1.657778 0.668557867828921
13 chr17:44777988:44778040:WNT3 -1.282798 0.61467112951709
14 chr4:122775166:122775266:TRPC3 -1.269060 0.61759501169366
15 chr3:60357325:60357413:FHIT -1.266671 0.610069267936541
Total deltapsi
1 0.139047439399753 -0.6472776
2 0.169115629694331 -0.5677637
3 0.313023025601665 -0.5636051
4 0.219649286101815 -0.5471778
5 0.231787036297737 -0.5317184
6 0.140656353549046 -0.5280372
7 0.224821665761922 -0.5223716
8 0.40129250694973 -0.5213804
9 0.34267701803463 -0.5185442
10 0.192803741815497 -0.5096807
11 0.171781437839461 -0.5059811
12 0.162584556335585 -0.5059733
13 0.109232403935245 -0.5054387
14 0.113169455110416 -0.5044256
15 0.110490691620035 -0.4995786It would be good to know the mean usage scores for these as well:
names=c("chr", "start","end","gene","strand", "peak", "meanUsage")
tot_usage=read.table("../data/PeakUsage_noMP/filtered_APApeaks_merged_allchrom_refseqGenes.TranscriptNoMP_sm_quant.Total_fixed.pheno.5percPeaks.txt", stringsAsFactors = F,col.names = names)
nuc_usage=read.table("../data/PeakUsage_noMP/filtered_APApeaks_merged_allchrom_refseqGenes.TranscriptNoMP_sm_quant.Nuclear_fixed.pheno.5percPeaks.txt",stringsAsFactors = F,col.names = names)- RGPD4:
Intronic Peak in the gene 64775-
Total mean usage: 0.1389744 Nuclear mean Usage: 0.7933333
The other peak in this gene is downstream of the annotated gene. 64776
Total mean Usage: 0.8610256 Nuclear mean Usage: 0.2066667
The nuclear has the truncated internal transcript.
- FAM86C1
The most used peak in this gene in the nuclear fraction is peak20610 it is chr11:71409928:71409980. This peak is upstream of the gene. This means the peak is probably not real. However this does not look like a misprimed read.
- SLC24A2
There are 2 peaks for this gene. They are downstream of the annoated in another gene but because they are on the opposite strand from the other gene, it makes sense for them to map to SLC24A2.
This means that without a strand specific protocol it would be difficult to see this effect.
Peak 118136 Total: 0.3012821 Nucelar: 0.8553846
Peak 118140 Total: 0.6987179 Nuclear: 0.1446154
The extended transcript is used in the nuclear.
- GALNTL5
This gene is close to the GALNT11 gene. They are also going in the same direction. The peak here is in the first intron of the GALNT11 gene. The other peak is way upsteream and looks like it is near the end of the PRKAG2-ASI gene chr7:151,570,858-151,585,995.
- NME2
Peak 50663
Total: 0.1615385 Nuclear:0.7335897
The other annotated peak is Peak 50662 Total:0.5564103 Nuclear:0.2407692
50663 is directly upstream of the UTR and 50662 is uspream in one of the introns.
- IFT22
This gene has 3 peaks in the filtered peaks however the one called here is upstream of the gene. The other two make a lot more sense:
109623: Total:0.1310256 Nuclear:0.63794872
109622:
Total: 0.1866667 Nuclear: 0.08897436
109621: Total:0.6371795 Nuclear: 0.23717949
From these results it looks like there is still differencial ussage in this gene. Peak 109621 is also significant with a delta PSI of 0.45275927.
- PCOLCE2
This peak is pretty far downstream of the gene but other genes in the region go the opposite way:
peak83037: Total:0.2151282 Nuclear: 0.7323077
peak83045: This peak is upstream of the UTR for the gene in one of the exons:
Total: 0.7848718 Nuclear: 0.2676923
These results look like the gene has significant run through in the nuclear fraciton.
- HNF1A
The 2 called peaks for this gene are upstream of the gene. Probably not actually correct. It does not look like any peaks in the track would work for this gene.
- WISP1
The peak is very far downstream but not in the wrong direciton for other peaks in the region. All of the peaks for this gene are pretty far downstream. I am not sure how best to know if these are real or not.
- FBXO22
The peak here is downstream of the gene close to the UTR of the next gene but because it goes the other direction, I can be confident in the assignemnt.
peak41217: Total: 0.16333333 Nuclear: 0.4917949
The other peaks in this gene are peak41206 in both fractions and low usage of peak41207 in total.
peak41206: Total: 0.58538462 Nuclear: 0.1776923
Again this looks like a case of run on read in the nuclear
- PCDH9
The peak is downsteam of the gene but it is in a region with many LINCs. This peak is 32303. THe other peaks for the gene are 32304 and 32305. Oeak 32305 is upstream meanin it is probably not real.
peak32303: Total:0.1761538 Nuclear: 0.6843590
Peak 32304: Total: 0.5946154 Nuclear: 0.2479487
This is another case of run on for the nuclear transcript.
CCM2L
The peak is downstream in on if the first exons of the HCK gene. It is difficult to know which you should assign the peak to because they are in the same orientation. All three of the called peaks for this gene are downstream in the HCK gene.WINT3:
The peak is downstream in the NSF gene but the NSF gene goes the other direction. Both peaks for this gene are downstream in the NSF gene.
Peak 50131
Total: 0.1087179 Nuclear: 0.5658974
Peak 50134
Total: 0.8912821 Nuclear: 0.4084615
Again here we see more run on for the nuclear fraction transcript.
- TRPC3
The peak here is in an intron of the downstreem gene BBS7. It is difficult to know which gene to assign the peak because they go the same direction.
peak89306 total: 0.1207692 nuclear:0.6010256
peak89307
total:0.8535897 nuclear: 0.3989744
Again here, if these peaks truly go with this gene, the nuclear transcript is the run on transcript.
- FHIT:
This peak is in an intron of the gene. This is also the only peak passing 5% in each fraction.
peak 79975:
Total:0.2602564
Nucelar:0.05923077
It is hard to draw further conclusions here.
I now want to look at some intermediate examples that are less likely to be outliers:
filterMidPSI=effectsize %>% filter(abs(deltapsi)>.1, abs(deltapsi)<.2) %>% arrange(deltapsi)
slice(filterMidPSI, 1:20) intron logef Nuclear
1 chr3:9824703:9824900:TADA3 -0.3663203 0.325432995242216
2 chr5:59173621:59173722:DEPDC1B -1.1379521 0.270435823995102
3 chr16:58746389:58746439:GOT2 -1.1858858 0.226137719840828
4 chr2:19534331:19534419:OSR1 -0.7480049 0.583655239822405
5 chr18:32824892:32825018:ZNF397 -0.8954891 0.332811386875754
6 chr5:27491617:27491709:CDH6 -0.3475314 0.340152540947897
7 chr12:26060628:26060675:BHLHE41 -1.1023581 0.284333101368843
8 chr8:126089041:126089194:WASHC5 -0.4613435 0.308742523793907
9 chr3:119762220:119762305:GPR156 -0.6754549 0.668378068472292
10 chr19:34702824:34702894:LSM14A -1.2379175 0.262806679986851
11 chr5:43482525:43482619:C5orf34 -0.5841307 0.237305958687913
12 chr7:17260895:17260936:AHR -1.0800012 0.257416122485577
13 chr7:98993610:98993692:ARPC1B -0.2835942 0.308021209679854
14 chr1:167612310:167612392:RCSD1 -0.4305542 0.291550619788812
15 chr7:5820254:5820322:OCM -0.4952238 0.779630820591706
16 chr16:3181668:3181753:ZSCAN10 -0.6372901 0.588452776814862
17 chr7:94260631:94260713:SGCE -0.9599485 0.279108870841054
18 chr19:47378498:47378583:FKRP -1.0682692 0.330580082811762
19 chr3:122481138:122481277:HSPBAP1 -0.4802691 0.433000666440278
20 chr3:12761795:12761880:TMEM40 -0.4885477 0.689878926943269
Total deltapsi
1 0.125571306501736 -0.1998617
2 0.0707522472721187 -0.1996836
3 0.0265447102065377 -0.1995930
4 0.384077797410421 -0.1995774
5 0.133435407762397 -0.1993760
6 0.14078100359469 -0.1993715
7 0.0850206759231001 -0.1993124
8 0.109466992933229 -0.1992755
9 0.469129223532137 -0.1992488
10 0.0636106463287395 -0.1991960
11 0.0381194363145641 -0.1991865
12 0.0583828866891181 -0.1990332
13 0.108998470216784 -0.1990227
14 0.0925532620325678 -0.1989974
15 0.580643826090524 -0.1989870
16 0.389493388354609 -0.1989594
17 0.0802511202513235 -0.1988578
18 0.131890177247821 -0.1986899
19 0.234320158423806 -0.1986805
20 0.491259391799099 -0.1986195- Tada3
This gene has 2 annotated UTRs. This peak is in the upstream 3’UTR.
Peak 7742:
Total:0.1382051 Nuclear: 0.29974359
The total only has 1 more annoated here: peak77240. This peak is in the downstream 3’ UTR
Total:0.7933333
Nucelar:0.47923077
The nuclear has 2 more upstream peaks here:
Peak77243:0.08589744 Peak77245:0.13461538
- DEPDC1B
This gene is close to PDE4D. The sig peak is in this gene. The 1 peak in the 3’ UTR of DEPDC1B is peak92690.
*GOT2
The internal peak for this gene is used less than 5% in the total and 22% in the nuclear. These are the only 2 peaks in our filtered set for the gene.
Peak45274: Total <5% Nuclear: 0.2202564
Peak45272: Total:0.9438462 Nuclear : 0.2202564
- OSR1
The 3 peaks for this gene are downstream of the gene with no other peaks in the region.
Peak 60538: Total:0.3035897 Nucelar: 0.5566667
The total fraction uses the one peak downstream at about the same rate.
- ZNF397
This gene also looks like it has multiple annotated UTRs.
Peak 53787
Total: 0.10461538
Nuclear: 0.22794872
This gene has 5 peaks and they run into the next gene downstream. Peaks 53807 and 53808 should probaby be assigned to ZNF271P but it is hard to tell.
- CDH6
These peaks should probably go to the LNC RNA LINC01021.
- BHLHE41
This doesnt make sense
- WASHC5
This is a good example. peak116684 is in an intron of the gene and peak116678 is at the end in UTR.
peak116684:
total:0.09589744 nuclear: 0.25256410
peak116678: total: 0.72538462 nuclear: 0.50384615
The other used peak in nucelar is peak116687 with 0.06641026.
- GPR156
The peaks are downstream in GSK3B.
- LSM14A
chr19:34702824:34702894
peak57649: This peak is in the intron of the gene.
Total: <5% Nuclear:0.16358974
In the total there are three peaks for this gene at over 5%.
peak57651 total:0.2943590 Nuclear:0.12179487
peak57655 Total:0.1356410 nuclear: 0.05461538
peak57656 total:0.2941026 nuclear: 0.10871795
Peaks 57655 adn 57656 are in the UTR.
This is a good example of internal PAS in the nuclear fraction.
C5orf34 chr5:43482525:43482619: peak912217. This peak is in the next gene downstream in the same direction. It is hard to tell which gene this goes with.
AHR: Peak 106198 This peak is upstream of the gene. Probably not ream chr7:17260895:17260936
However 106205 is an interesting peak here. It is not used at 5% in total but it used at almost 17% in nuclear.
- ARPC1B chr7:98993610:98993692 This is peak 109344. It is downstream in PDAP1 but also looks like its still the annoataed UTR for the ARPC1B gene . But this gene is the other direction.
peak 109344: total:0.1546154 nuclear: 0.27615385
The nuclear has 6 peaks at > 5% for this gene and total only has 2. The most used total is peak109343. But it does not look like that in IGV. It looks like it uses 109344.
- RCSD1
peak 8281
total: 0.08051282 nuclear: 0.21512821
peak8293 is the other used peak in total. It is in the annoated 3’ UTR
total:0.67102564 nuclear:0.33769231
There are 2 more peaks also used at moderate levels here.
OCM Peak is upstream. Not correct
ZSCAN10 peak is upstream. not correct
SGCE
peak 190201 In an intron of the gene. total:0.10589744 nuclear: 0.2587179
The utr peak is 109198
total:0.58205128 nuclear:0.3894872
- FKRB
chr19:47378498:47378583
Looks like there must be a transcribed element here but thereis not an annotated gene. FKRB is pretty far upstream.
- HSPBAP1
peak81962 total:0.26000000 nuclear: 0.41871795
the most used total peak is 81961 it is in the 3’ UTR
total: 0.64743590 nuclear:0.49000000
- TMEM40
peak77434. The peaks for this gene are all outside. It is a bit confusing. 77435 is upstream so this is probably not correct
Conclusions:
Gene to peak annotations are difficult, especially when the genes are in the same direction close to each other
We need to make sure peaks cannot be upstream of the gene.
We need to add LINC to the annotation
Session information
sessionInfo()R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS 10.14.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] bindrcpp_0.2.2 cowplot_0.9.3 reshape2_1.4.3 forcats_0.3.0
[5] stringr_1.3.1 dplyr_0.7.6 purrr_0.2.5 readr_1.1.1
[9] tidyr_0.8.1 tibble_1.4.2 ggplot2_3.0.0 tidyverse_1.2.1
[13] workflowr_1.1.1
loaded via a namespace (and not attached):
[1] tidyselect_0.2.4 haven_1.1.2 lattice_0.20-35
[4] colorspace_1.3-2 htmltools_0.3.6 yaml_2.2.0
[7] rlang_0.2.2 R.oo_1.22.0 pillar_1.3.0
[10] glue_1.3.0 withr_2.1.2 R.utils_2.7.0
[13] modelr_0.1.2 readxl_1.1.0 bindr_0.1.1
[16] plyr_1.8.4 munsell_0.5.0 gtable_0.2.0
[19] cellranger_1.1.0 rvest_0.3.2 R.methodsS3_1.7.1
[22] evaluate_0.11 knitr_1.20 broom_0.5.0
[25] Rcpp_0.12.19 scales_1.0.0 backports_1.1.2
[28] jsonlite_1.5 hms_0.4.2 digest_0.6.17
[31] stringi_1.2.4 grid_3.5.1 rprojroot_1.3-2
[34] cli_1.0.1 tools_3.5.1 magrittr_1.5
[37] lazyeval_0.2.1 crayon_1.3.4 whisker_0.3-2
[40] pkgconfig_2.0.2 xml2_1.2.0 lubridate_1.7.4
[43] assertthat_0.2.0 rmarkdown_1.10 httr_1.3.1
[46] rstudioapi_0.8 R6_2.3.0 nlme_3.1-137
[49] git2r_0.23.0 compiler_3.5.1
This reproducible R Markdown analysis was created with workflowr 1.1.1