Create Gviz genome track plots
Briana Mittleman
2017-12-13
Last updated: 2017-12-13
Code version: 3ed7074
The goal of this analysis is to create nice plots showing that we are getting as much information as the 1 lane from the Mayer sample. I will do this with our merged data vs. their 1 lane.
Genes from IGV that I want to use:
HERPUD1 chr16:56,964,002-56,979,793
ACTB chr7:5,564,779-5,572,232
CCNB2 chr15:59,396,707-59,401,006
chr11:234,336-239,997
KIAA0100 chr17:26,968,078-26,974,887
HECTD1 chr14:31,672,040-31,681,043
STAG1 chr3:136,469,421-136,472,771
Load Packages:
library(Gviz)Loading required package: S4VectorsLoading required package: stats4Loading required package: BiocGenericsLoading required package: parallel
Attaching package: 'BiocGenerics'The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLBThe following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, cbind, colMeans,
colnames, colSums, do.call, duplicated, eval, evalq, Filter,
Find, get, grep, grepl, intersect, is.unsorted, lapply,
lengths, Map, mapply, match, mget, order, paste, pmax,
pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce,
rowMeans, rownames, rowSums, sapply, setdiff, sort, table,
tapply, union, unique, unsplit, which, which.max, which.min
Attaching package: 'S4Vectors'The following object is masked from 'package:base':
expand.gridLoading required package: IRangesLoading required package: GenomicRangesLoading required package: GenomeInfoDbLoading required package: gridlibrary(GenomicRanges)
library(biomaRt)Upload data:
chr= "chr7"
gen= "hg19"
options(ucscChromosomeNames=FALSE)
merged_data_7= DataTrack(range = "../data/merged_Net1.bam", genome = gen, type = "h", name = "Merged", window = -1, chromosome = "7")
mayer_data_7= DataTrack(range = "../data/SRR1575922-sort.bam", genome = gen, type = "h", name = "Mayer", window = -1, chromosome = "7")
data("geneModels")
grtrack= GeneRegionTrack(geneModels, genome = gen, chromosome = chr, name = "Gene Model")
itrack= IdeogramTrack(genome = gen, chromosome = chr)
gtrack = GenomeAxisTrack()
#plots
plotTracks(list(gtrack,merged_data_7, mayer_data_7), from = 5564779, to = 5572232, background.title="darkblue", background.panel = "#FFFEDB")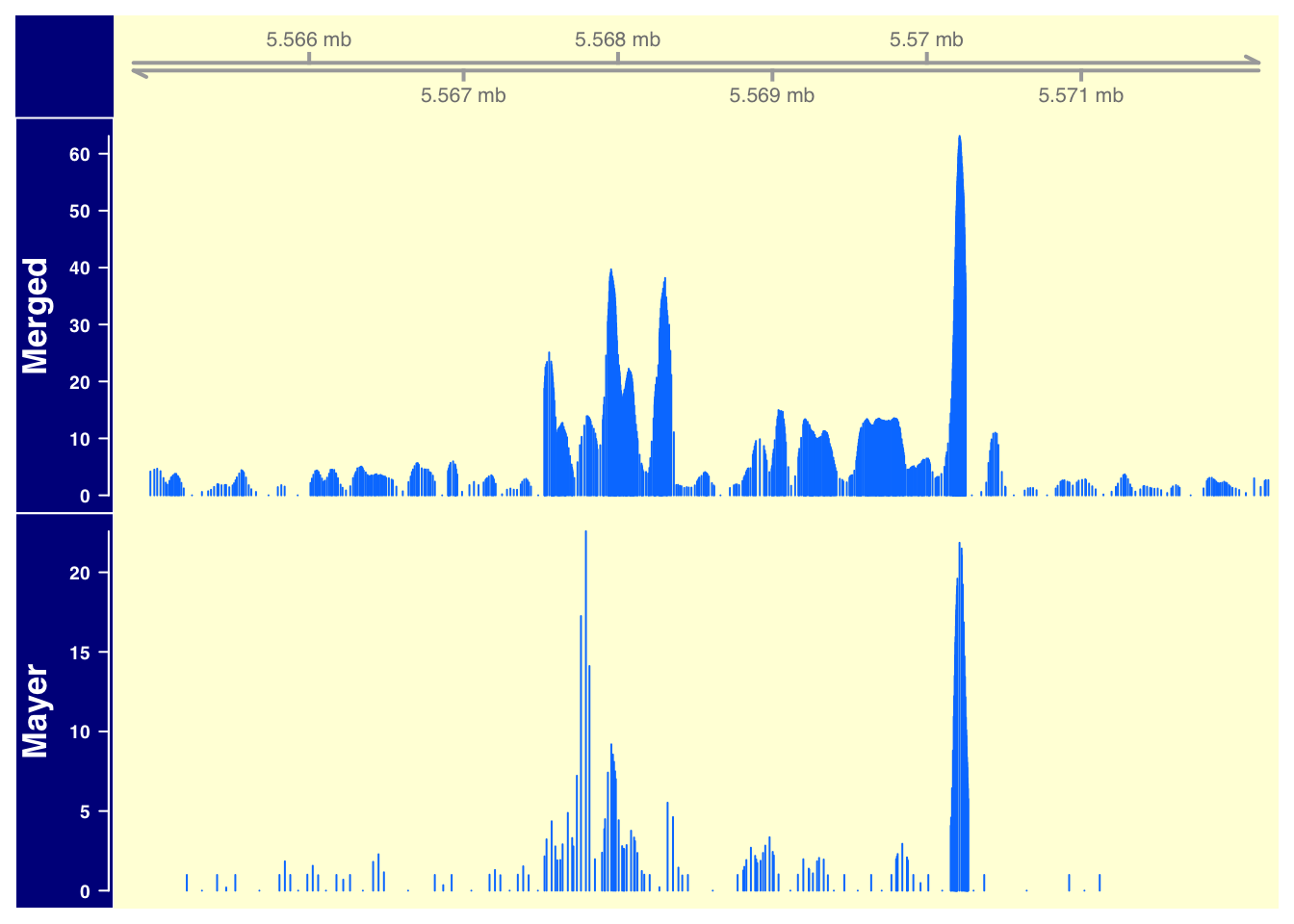
plotTracks(list(itrack, grtrack), transcriptAnnotation= "symbol", background.title="blue")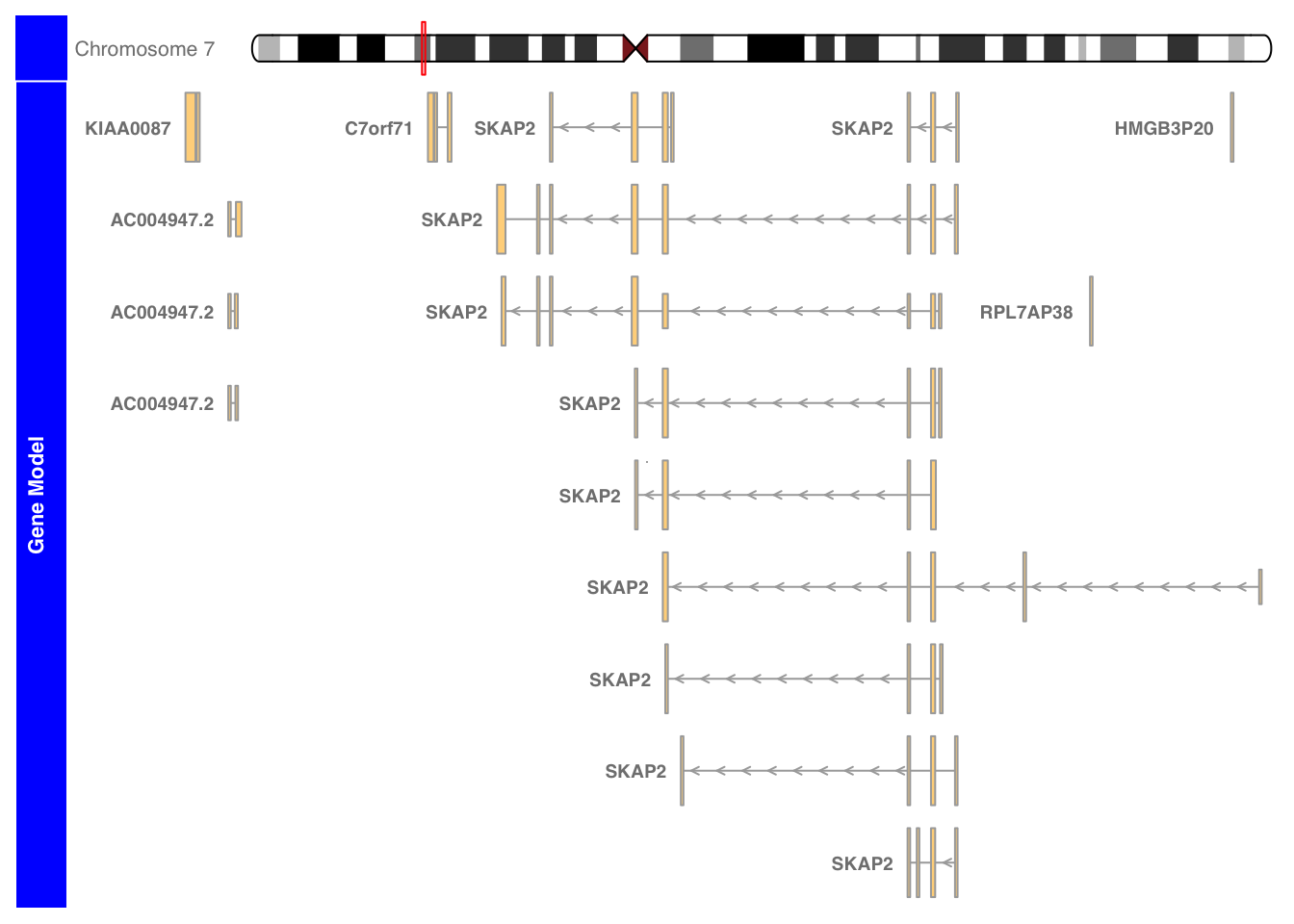
merged_anno_7= AnnotationTrack(range = "../data/merged_Net1.bam", genome = gen, name = "Merged", window = -1, chromosome = "7")
mayer_anno_7= AnnotationTrack(range = "../data/SRR1575922-sort.bam", genome = gen, name = "Mayer", window = -1, chromosome = "7")
plotTracks(list(merged_data_7, merged_anno_7), from = 5564779, to = 5572232)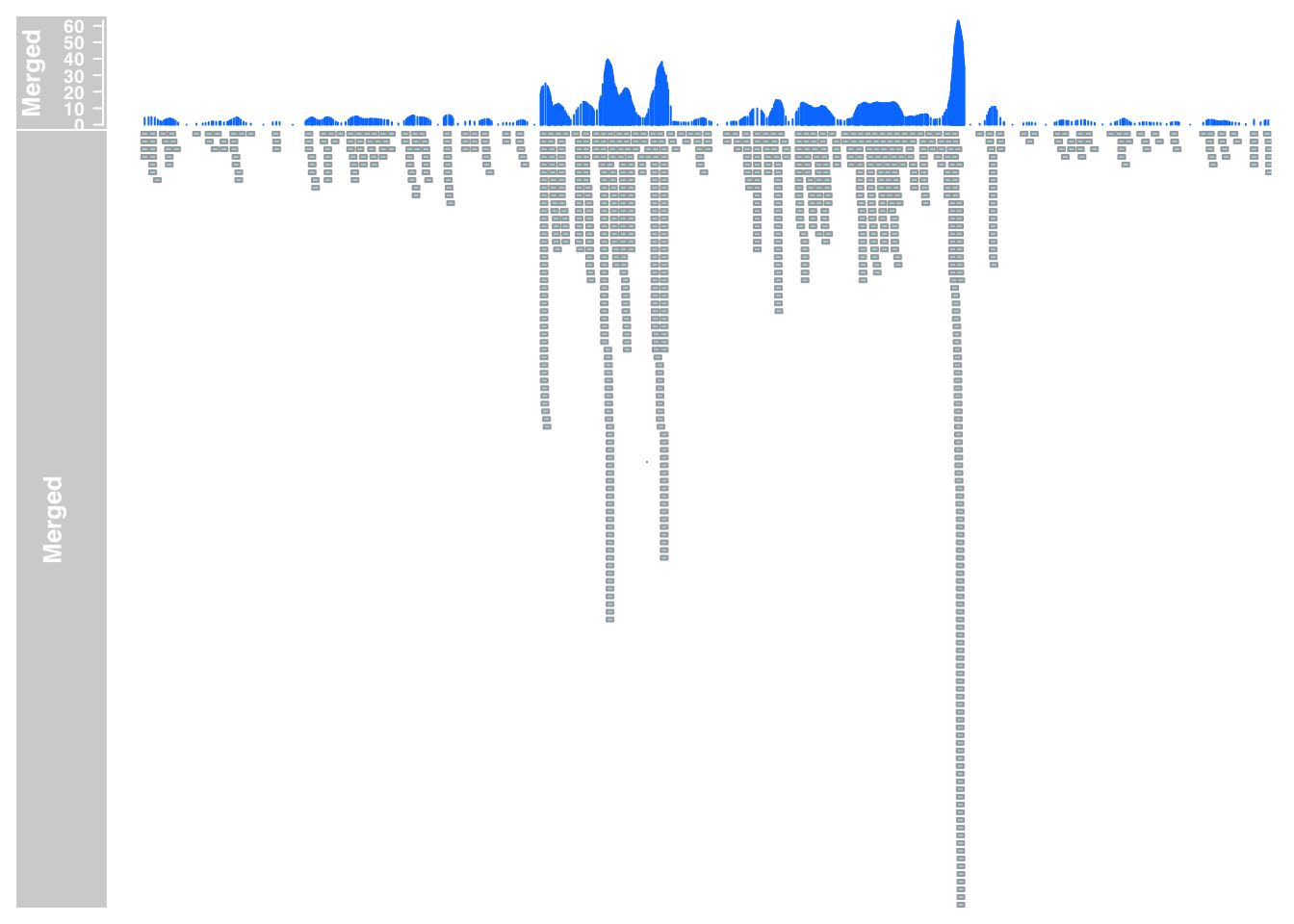
plotTracks(list(mayer_data_7,mayer_anno_7), from = 5564779, to = 5572232)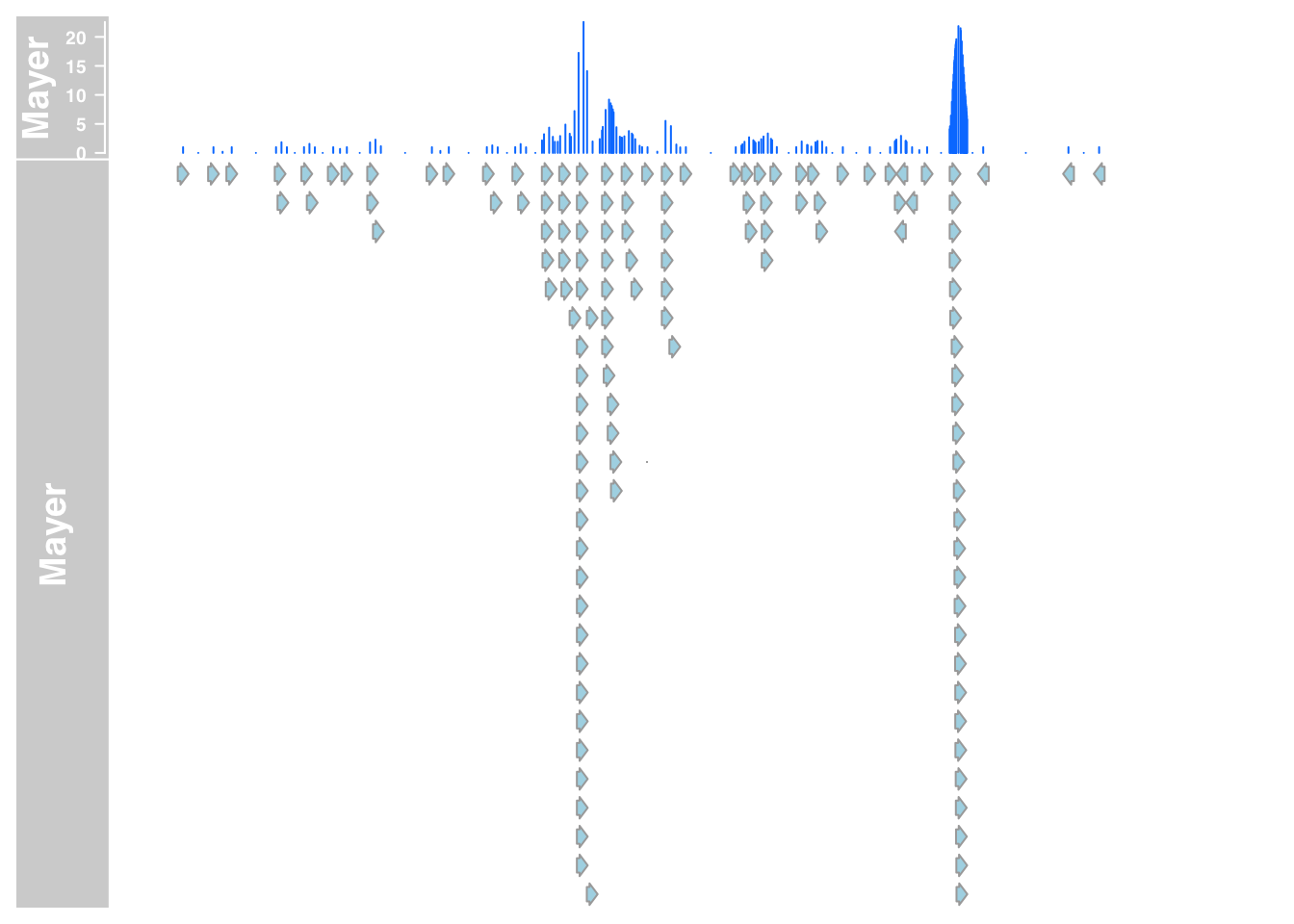
Session information
sessionInfo()R version 3.4.2 (2017-09-28)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] biomaRt_2.34.0 Gviz_1.22.1 GenomicRanges_1.30.0
[4] GenomeInfoDb_1.14.0 IRanges_2.12.0 S4Vectors_0.16.0
[7] BiocGenerics_0.24.0
loaded via a namespace (and not attached):
[1] ProtGenerics_1.10.0 bitops_1.0-6
[3] matrixStats_0.52.2 bit64_0.9-7
[5] RColorBrewer_1.1-2 progress_1.1.2
[7] httr_1.3.1 rprojroot_1.2
[9] tools_3.4.2 backports_1.1.1
[11] R6_2.2.2 rpart_4.1-11
[13] Hmisc_4.0-3 DBI_0.7
[15] lazyeval_0.2.1 colorspace_1.3-2
[17] nnet_7.3-12 gridExtra_2.3
[19] prettyunits_1.0.2 RMySQL_0.10.13
[21] bit_1.1-12 curl_3.0
[23] compiler_3.4.2 git2r_0.19.0
[25] Biobase_2.38.0 htmlTable_1.11.0
[27] DelayedArray_0.4.1 rtracklayer_1.38.2
[29] scales_0.5.0 checkmate_1.8.5
[31] stringr_1.2.0 digest_0.6.12
[33] Rsamtools_1.30.0 foreign_0.8-69
[35] rmarkdown_1.8 XVector_0.18.0
[37] base64enc_0.1-3 dichromat_2.0-0
[39] pkgconfig_2.0.1 htmltools_0.3.6
[41] ensembldb_2.2.0 BSgenome_1.46.0
[43] htmlwidgets_0.9 rlang_0.1.4
[45] rstudioapi_0.7 RSQLite_2.0
[47] BiocInstaller_1.28.0 shiny_1.0.5
[49] bindr_0.1 BiocParallel_1.12.0
[51] acepack_1.4.1 dplyr_0.7.4
[53] VariantAnnotation_1.24.2 RCurl_1.95-4.8
[55] magrittr_1.5 GenomeInfoDbData_0.99.1
[57] Formula_1.2-2 Matrix_1.2-12
[59] Rcpp_0.12.14 munsell_0.4.3
[61] stringi_1.1.6 yaml_2.1.15
[63] SummarizedExperiment_1.8.0 zlibbioc_1.24.0
[65] plyr_1.8.4 AnnotationHub_2.10.1
[67] blob_1.1.0 lattice_0.20-35
[69] Biostrings_2.46.0 splines_3.4.2
[71] GenomicFeatures_1.30.0 knitr_1.17
[73] XML_3.98-1.9 glue_1.2.0
[75] evaluate_0.10.1 biovizBase_1.26.0
[77] latticeExtra_0.6-28 data.table_1.10.4-3
[79] httpuv_1.3.5 gtable_0.2.0
[81] purrr_0.2.4 tidyr_0.7.2
[83] assertthat_0.2.0 ggplot2_2.2.1
[85] mime_0.5 xtable_1.8-2
[87] AnnotationFilter_1.2.0 survival_2.41-3
[89] tibble_1.3.4 GenomicAlignments_1.14.1
[91] AnnotationDbi_1.40.0 memoise_1.1.0
[93] bindrcpp_0.2 cluster_2.0.6
[95] interactiveDisplayBase_1.16.0This R Markdown site was created with workflowr