Explore UMI Usage in Netseq1 libary
Briana Mittleman
2017-11-08
Last updated: 2017-11-13
Code version: a25b69a
In this analysis I will explore the UMI usage in the Net-Seq1 library. Due to low read counts in the total sample, I will exclude this sample from the analysis.
This code is used to create a text file that I can explore in R. It has a list of all of the UMIs used for the sample sorted by usage with the number of times each is used. This is run before the duduplication step.
samtools view {file} | tr "_" "\t" | cut -f 2 | sort | uniq -c > ../../output/UMI_{file}_stat.txtPackages
library("tidyr")
library("dplyr")
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionlibrary("ggplot2")
library("seqLogo")Loading required package: gridlibrary("Biostrings")Loading required package: BiocGenericsLoading required package: parallel
Attaching package: 'BiocGenerics'The following objects are masked from 'package:parallel':
clusterApply, clusterApplyLB, clusterCall, clusterEvalQ,
clusterExport, clusterMap, parApply, parCapply, parLapply,
parLapplyLB, parRapply, parSapply, parSapplyLBThe following objects are masked from 'package:dplyr':
combine, intersect, setdiff, unionThe following objects are masked from 'package:stats':
IQR, mad, sd, var, xtabsThe following objects are masked from 'package:base':
anyDuplicated, append, as.data.frame, cbind, colMeans,
colnames, colSums, do.call, duplicated, eval, evalq, Filter,
Find, get, grep, grepl, intersect, is.unsorted, lapply,
lengths, Map, mapply, match, mget, order, paste, pmax,
pmax.int, pmin, pmin.int, Position, rank, rbind, Reduce,
rowMeans, rownames, rowSums, sapply, setdiff, sort, table,
tapply, union, unique, unsplit, which, which.max, which.minLoading required package: S4VectorsLoading required package: stats4
Attaching package: 'S4Vectors'The following objects are masked from 'package:dplyr':
first, renameThe following object is masked from 'package:tidyr':
expandThe following object is masked from 'package:base':
expand.gridLoading required package: IRanges
Attaching package: 'IRanges'The following objects are masked from 'package:dplyr':
collapse, desc, sliceLoading required package: XVector
Attaching package: 'Biostrings'The following object is masked from 'package:base':
strsplitrequire("Biostrings")Input data
prepare_UMI_data=function(path.txt){
x=read.delim(file=path.txt, header = FALSE,stringsAsFactors = FALSE)
colnames(x) <- "UMI"
x= data.frame(sapply(x, trimws), stringsAsFactors = FALSE)
x= separate(data=x, col = UMI, into= c("number", "umi"), sep="\\s+")
x$number= as.numeric(x$number)
x= arrange(x, desc(number))
return(x)
}
UMI_18486_dep = prepare_UMI_data("../data/UMI_18486_dep_stat.txt")
UMI_18508_dep= prepare_UMI_data("../data/UMI_18508_dep_stat.txt")
UMI_18508_nondep= prepare_UMI_data("../data/UMI_18508_nondep_stat.txt")
UMI_19238_dep= prepare_UMI_data("../data/UMI_19238_dep_stat.txt")
UMI_mayer= prepare_UMI_data("../data/UMI_mayer_stat.txt")Plot the umi distributions
par(mfrow = c(2,3))
plot(UMI_18486_dep$number, ylab="UMI count", xlab="UMI", main="18486-dep distribution")
plot(UMI_18508_dep$number, ylab="UMI count", xlab="UMI", main="18508-dep distribution")
plot(UMI_18508_nondep$number, ylab="UMI count", xlab="UMI", main="1508- nondep distribution")
plot(UMI_19238_dep$number, ylab="UMI count", xlab="UMI", main="19238-dep distribution")
plot(UMI_mayer$number, ylab="UMI count", xlab="UMI", main="Mayer distribution")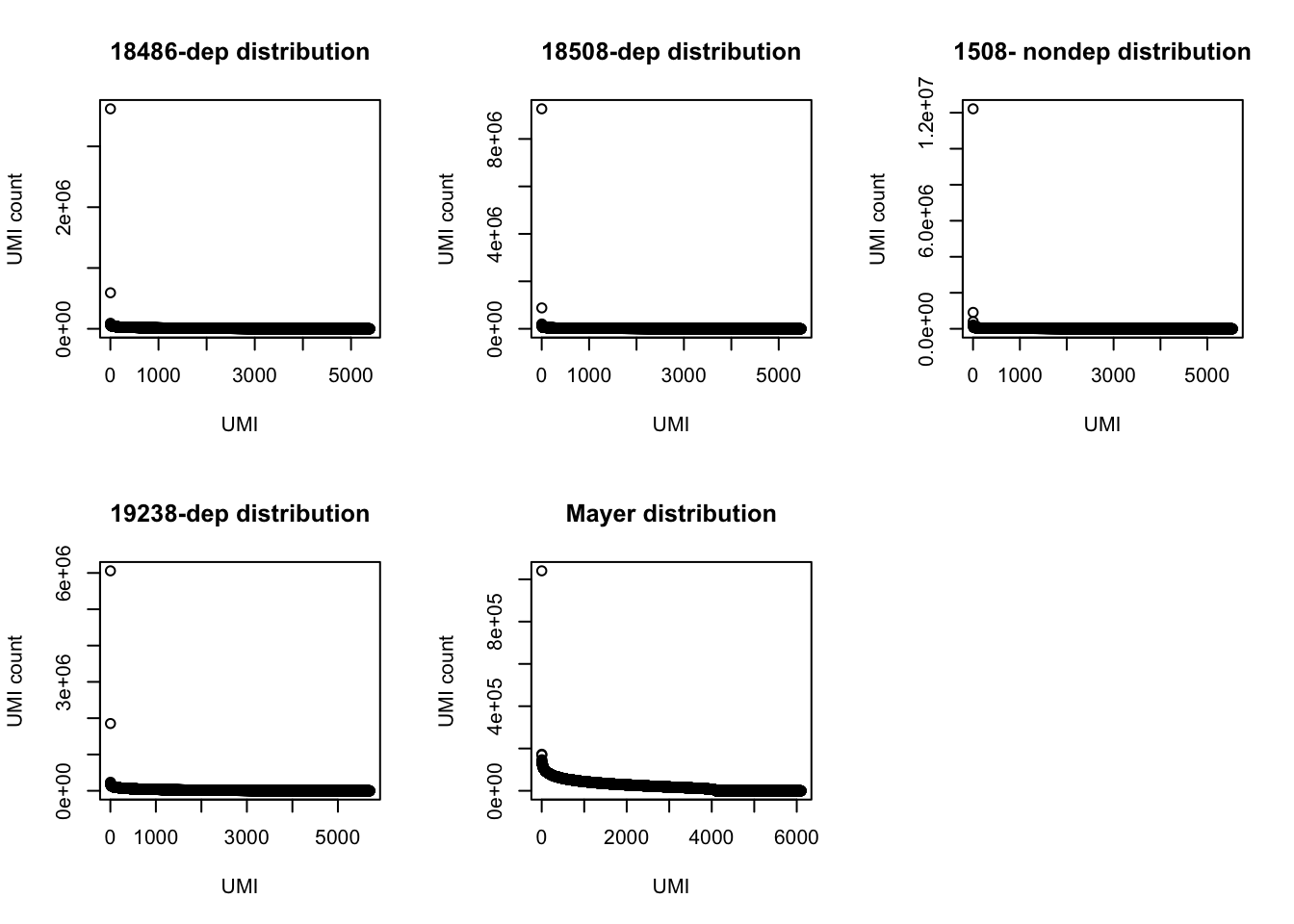 Look at the top used UMI for each data set.
Look at the top used UMI for each data set.
UMI_18486_dep[1:3,] number umi
1 3617979 ATCTCG
2 592512 CACCCG
3 90128 TCTCGTUMI_18508_dep[1:3,] number umi
1 9270083 ATCTCG
2 880379 CACCCG
3 201796 TCTCGTUMI_18508_nondep[1:3, ] number umi
1 12216803 ATCTCG
2 911426 CACCCG
3 401897 TCTCGTUMI_19238_dep[1:3,] number umi
1 6058977 ATCTCG
2 1852855 CACCCG
3 235866 TATCTCUMI_mayer[1:3,] number umi
1 1040195 ATCTCG
2 172910 TTTCAC
3 169350 TTACACThe top used UMIs are similar accross samples. This preference could be due to annealing temperatures.(Conversation with Po) All data sets show an overrepresentation of a few UMIs, I will remove the top 5 to get a better look at the distribution.
par(mfrow = c(2,3))
plot(UMI_18486_dep[6:5388,]$number, ylab="UMI count", xlab="UMI", main="18486-dep distribution -5")
plot(UMI_18508_dep[6:5471,]$number, ylab="UMI count", xlab="UMI", main="18508-dep distribution -5")
plot(UMI_18508_nondep[6:5535,]$number, ylab="UMI count", xlab="UMI", main="18508-nondep distribution -5")
plot(UMI_19238_dep[6:5699,]$number, ylab="UMI count", xlab="UMI", main="19328-dep distribution -5")
plot(UMI_mayer[6:6101,]$number, ylab="UMI count", xlab="UMI", main="Mayer distribution -5")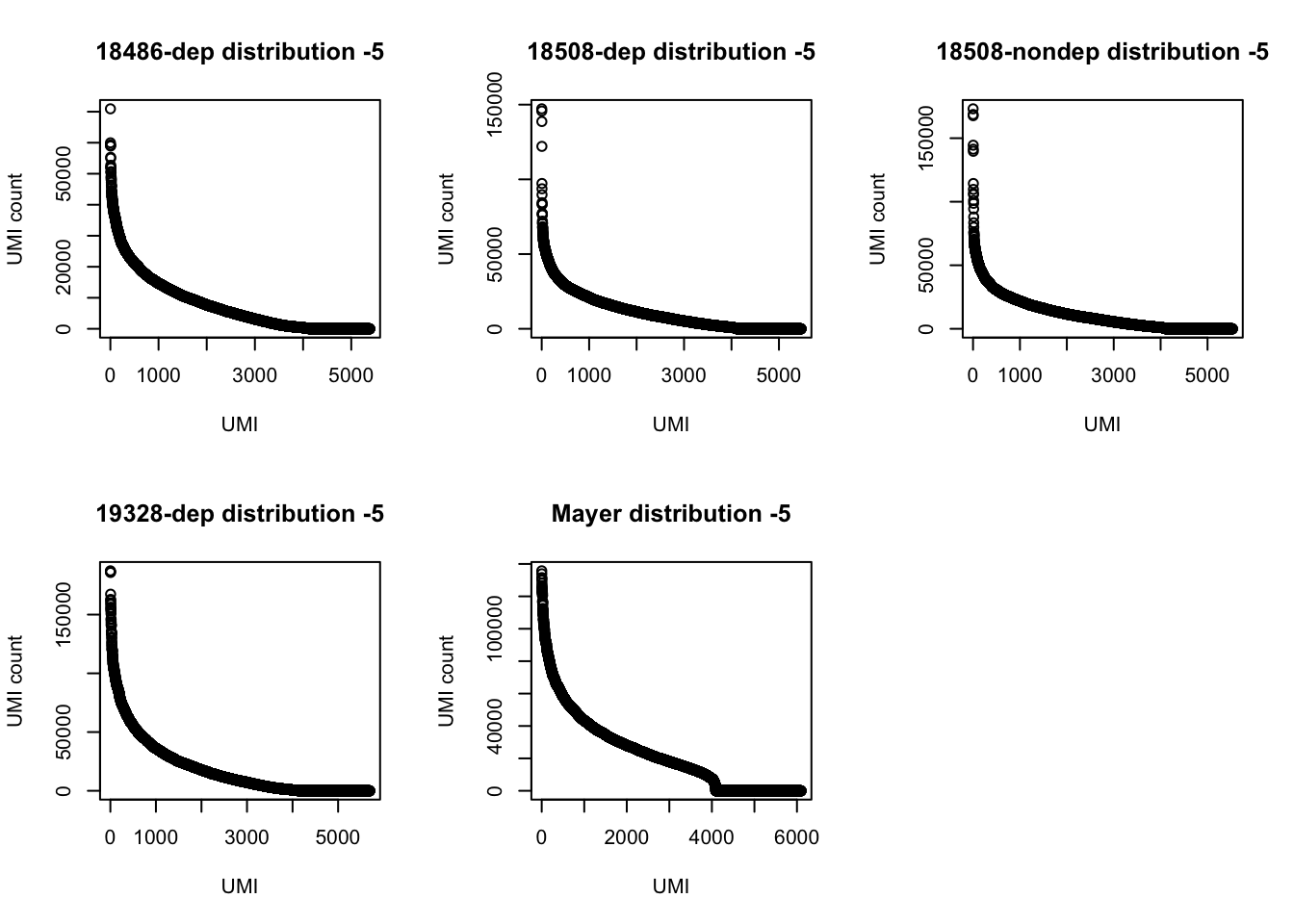
Seq Logo Plots
Use Biostrings to get the PMW then create the logoplots with seqlogo.
#source("https://bioconductor.org/biocLite.R")
#biocLite("seqLogo")
#source("http://bioconductor.org/biocLite.R")
#biocLite("Biostrings")
#set= DNAStringSet(UMI_18486_dep$umi)
#length(set)
#set.freq=data.frame(alphabetFrequency(set, baseOnly=T, as.prob=T))
#set_noN=set[set.freq$other==0,]
#length(set_noN)
#width(set_noN)
#x=consensusMatrix(set_noN) #problem here, getting 1024 for all
#freq_18486= PWM(x[1:4,])
#sum(UMI_18486_dep$number==0) > 0 : shows no UMIs are never used
#seqLogo(freq_18486, ic.scale = TRUE, xaxis = TRUE, yaxis = TRUE, xfontsize = 15, yfontsize =15)Try with a different package:
#library("devtools")
#install_github("omarwagih/ggseqlogo")
require(ggseqlogo)Loading required package: ggseqlogocs1 = make_col_scheme(chars=c('A', 'T', 'C', 'G', 'N'), groups=c('A', 'T', 'C', 'G', 'N'), cols=c('red', 'blue', 'green', 'yellow', 'pink'))
par(mfrow = c(2,3))
ggseqlogo(UMI_18486_dep$umi, col_scheme=cs1)
ggseqlogo(UMI_18508_dep$umi, col_scheme=cs1)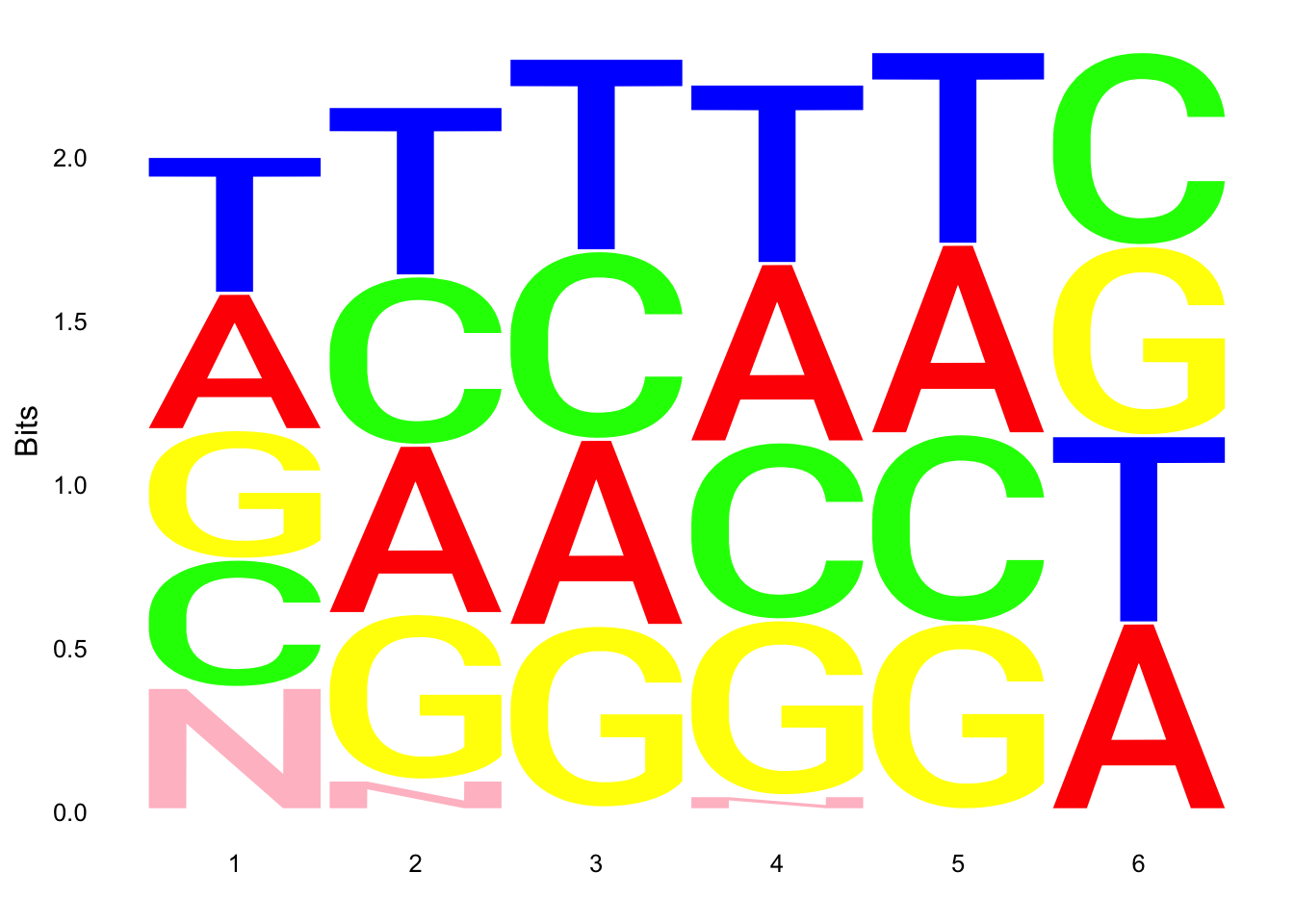
ggseqlogo(UMI_18508_nondep$umi, col_scheme=cs1)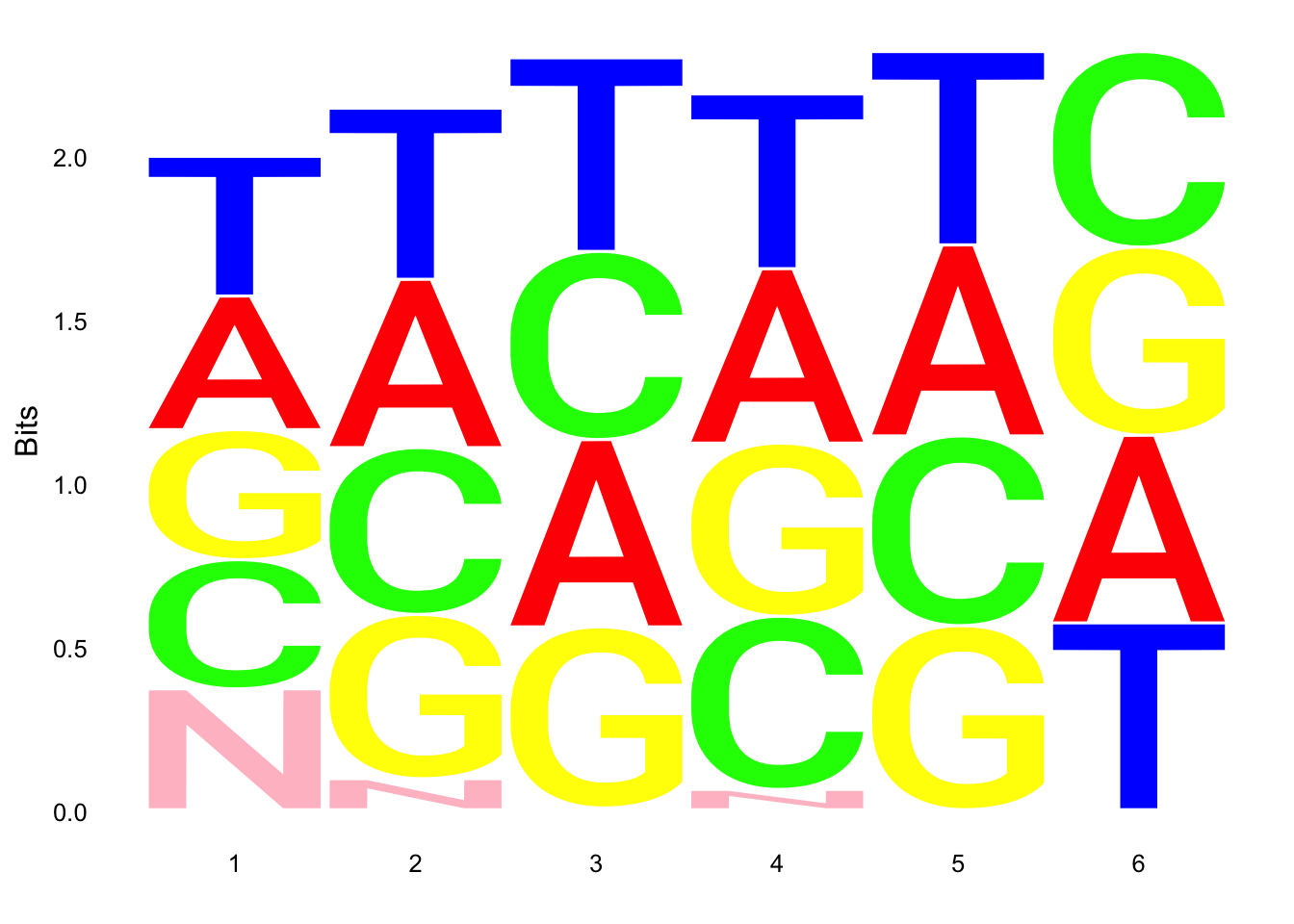
ggseqlogo(UMI_19238_dep$umi, col_scheme=cs1)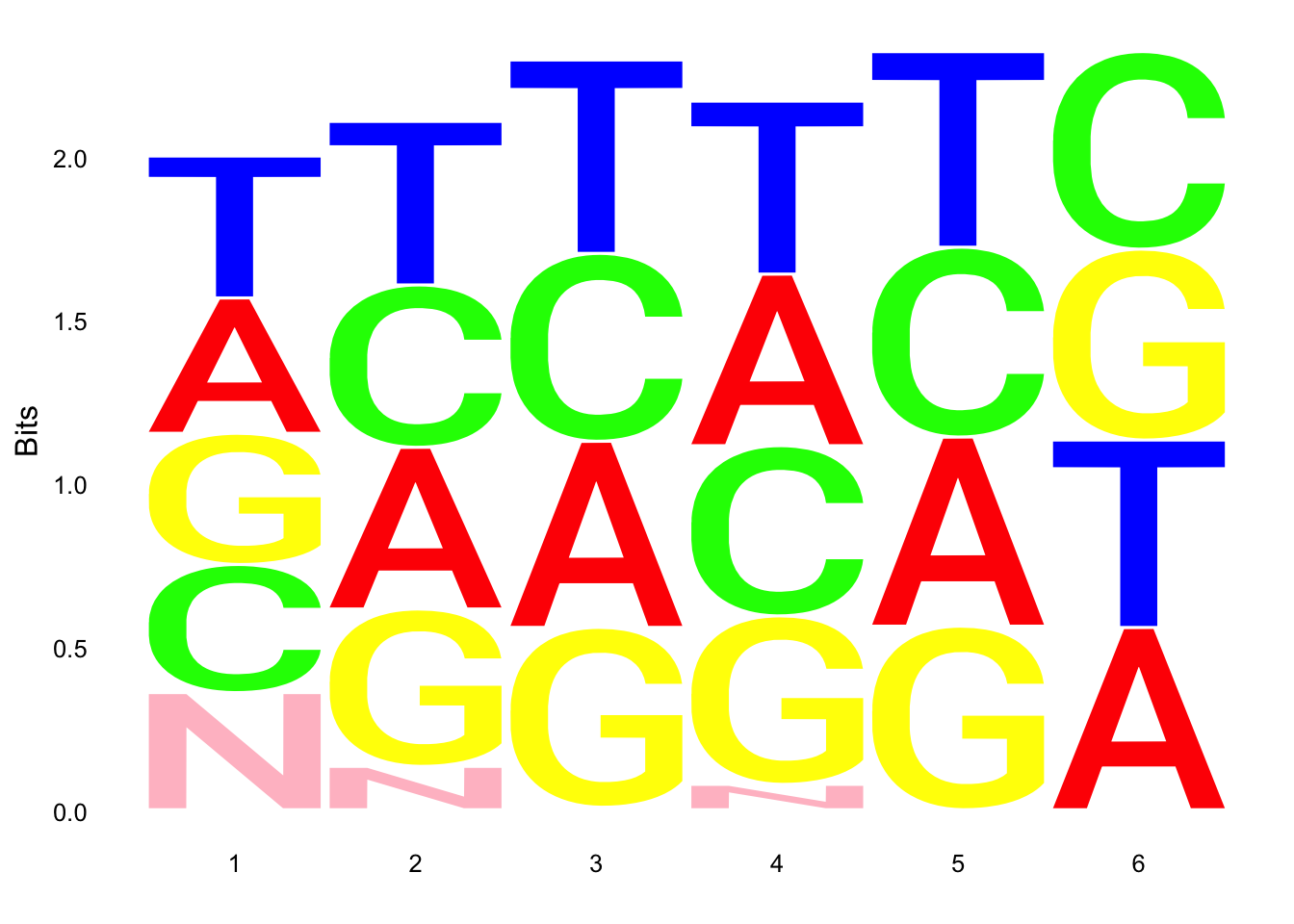
ggseqlogo(UMI_mayer$umi, col_scheme=cs1) Does not look like we get overrepresentation of one letter at any particular location in the UMI.
Does not look like we get overrepresentation of one letter at any particular location in the UMI.
test.seqs= c("ATGC", "TAGC", "ATGC", "ATGC")
ggseqlogo(test.seqs, col_scheme=cs1)
sessionInfo()R version 3.4.2 (2017-09-28)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS Sierra 10.12.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats4 parallel grid stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] ggseqlogo_0.1 bindrcpp_0.2 Biostrings_2.46.0
[4] XVector_0.18.0 IRanges_2.12.0 S4Vectors_0.16.0
[7] BiocGenerics_0.24.0 seqLogo_1.44.0 ggplot2_2.2.1
[10] dplyr_0.7.4 tidyr_0.7.2
loaded via a namespace (and not attached):
[1] Rcpp_0.12.13 compiler_3.4.2 git2r_0.19.0 plyr_1.8.4
[5] bindr_0.1 tools_3.4.2 zlibbioc_1.24.0 digest_0.6.12
[9] evaluate_0.10.1 tibble_1.3.4 gtable_0.2.0 pkgconfig_2.0.1
[13] rlang_0.1.4 yaml_2.1.14 stringr_1.2.0 knitr_1.17
[17] tidyselect_0.2.3 rprojroot_1.2 glue_1.2.0 R6_2.2.2
[21] rmarkdown_1.6 purrr_0.2.4 magrittr_1.5 backports_1.1.1
[25] scales_0.5.0 htmltools_0.3.6 assertthat_0.2.0 colorspace_1.3-2
[29] labeling_0.3 stringi_1.1.5 lazyeval_0.2.1 munsell_0.4.3 This R Markdown site was created with workflowr