voom_limma_weight_change
Lauren Blake
2018-08-20
Last updated: 2018-08-28
workflowr checks: (Click a bullet for more information)-
✖ R Markdown file: uncommitted changes
The R Markdown is untracked by Git. To know which version of the R Markdown file created these results, you’ll want to first commit it to the Git repo. If you’re still working on the analysis, you can ignore this warning. When you’re finished, you can runwflow_publishto commit the R Markdown file and build the HTML. -
✔ Environment: empty
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
-
✔ Seed:
set.seed(12345)The command
set.seed(12345)was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible. -
✔ Session information: recorded
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
-
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.✔ Repository version: 241c630
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can usewflow_publishorwflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.Ignored files: Ignored: .DS_Store Ignored: analysis/.DS_Store Ignored: data/.DS_Store Ignored: data/aux_info/ Ignored: data/hg_38/ Ignored: data/libParams/ Ignored: output/.DS_Store Untracked files: Untracked: _workflowr.yml Untracked: analysis/Collection_dates.Rmd Untracked: analysis/Converting_IDs.Rmd Untracked: analysis/Global_variation.Rmd Untracked: analysis/Preliminary_clinical_covariate.Rmd Untracked: analysis/VennDiagram2018-07-24_06-55-46.log Untracked: analysis/VennDiagram2018-07-24_06-56-13.log Untracked: analysis/VennDiagram2018-07-24_06-56-50.log Untracked: analysis/VennDiagram2018-07-24_06-58-41.log Untracked: analysis/VennDiagram2018-07-24_07-00-07.log Untracked: analysis/VennDiagram2018-07-24_07-00-42.log Untracked: analysis/VennDiagram2018-07-24_07-01-08.log Untracked: analysis/VennDiagram2018-08-17_15-13-24.log Untracked: analysis/VennDiagram2018-08-17_15-13-30.log Untracked: analysis/VennDiagram2018-08-17_15-15-06.log Untracked: analysis/VennDiagram2018-08-17_15-16-01.log Untracked: analysis/VennDiagram2018-08-17_15-17-51.log Untracked: analysis/VennDiagram2018-08-17_15-18-42.log Untracked: analysis/VennDiagram2018-08-17_15-19-21.log Untracked: analysis/VennDiagram2018-08-20_09-07-57.log Untracked: analysis/VennDiagram2018-08-20_09-08-37.log Untracked: analysis/VennDiagram2018-08-26_19-54-03.log Untracked: analysis/VennDiagram2018-08-26_20-47-08.log Untracked: analysis/VennDiagram2018-08-26_20-49-49.log Untracked: analysis/VennDiagram2018-08-27_00-04-36.log Untracked: analysis/VennDiagram2018-08-27_00-09-27.log Untracked: analysis/VennDiagram2018-08-27_00-13-57.log Untracked: analysis/VennDiagram2018-08-27_00-16-32.log Untracked: analysis/VennDiagram2018-08-27_10-00-25.log Untracked: analysis/VennDiagram2018-08-28_06-03-13.log Untracked: analysis/VennDiagram2018-08-28_06-03-14.log Untracked: analysis/VennDiagram2018-08-28_06-05-50.log Untracked: analysis/VennDiagram2018-08-28_06-06-58.log Untracked: analysis/VennDiagram2018-08-28_06-10-12.log Untracked: analysis/VennDiagram2018-08-28_06-10-13.log Untracked: analysis/VennDiagram2018-08-28_06-18-29.log Untracked: analysis/VennDiagram2018-08-28_07-22-26.log Untracked: analysis/VennDiagram2018-08-28_07-22-27.log Untracked: analysis/background_dds_david.csv Untracked: analysis/correlations_bet_covariates.Rmd Untracked: analysis/correlations_over_time.Rmd Untracked: analysis/genocode_annotation_info.Rmd Untracked: analysis/genotypes.Rmd Untracked: analysis/import_transcript_level_estimates.Rmd Untracked: analysis/test_dds_david.csv Untracked: analysis/variables_by_time.Rmd Untracked: analysis/voom_limma.Rmd Untracked: analysis/voom_limma_hg37.Rmd Untracked: analysis/voom_limma_weight_change.Rmd Untracked: data/BAN2 Dates_T1_T2.xlsx Untracked: data/BAN_DATES.csv Untracked: data/BAN_DATES.xlsx Untracked: data/BAN_DATES_txt.csv Untracked: data/Ban_geno.csv Untracked: data/Ban_geno.xlsx Untracked: data/Blood_dates.txt Untracked: data/DAVID_background.txt Untracked: data/DAVID_list_T1T2.txt Untracked: data/DAVID_list_T1T2_weight.txt Untracked: data/DAVID_list_T2T3.txt Untracked: data/DAVID_list_T2T3_weight.txt Untracked: data/DAVID_results/ Untracked: data/DAVID_top100_list_T1T2.txt Untracked: data/DAVID_top100_list_T1T2_weight.txt Untracked: data/DAVID_top100_list_T2T3.txt Untracked: data/DAVID_top100_list_T2T3_weight.txt Untracked: data/Eigengenes/ Untracked: data/FemaleWeightRestoration-01-dataInput.RData Untracked: data/FemaleWeightRestoration-resid-01-dataInput.RData Untracked: data/FemaleWeightRestoration-resid-T1T2-01-dataInput.RData Untracked: data/HTSF_IDs.sav Untracked: data/Homo_sapiens.GRCh38.v22_table.txt Untracked: data/Labels.csv Untracked: data/Labels.xlsx Untracked: data/RIN.xlsx Untracked: data/RIN_over_time.csv Untracked: data/RIN_over_time.xlsx Untracked: data/T0_consolid.csv Untracked: data/T0_consolid.xlsx Untracked: data/age_t1.txt Untracked: data/birthday_age.csv Untracked: data/birthday_age.xlsx Untracked: data/clinical_sample_info.csv Untracked: data/clinical_sample_info_geno.csv Untracked: data/cmd_info.json Untracked: data/counts_hg37_gc_txsalmon.RData Untracked: data/counts_hg38_gc.RData Untracked: data/counts_hg38_gc_dds.RData Untracked: data/counts_hg38_gc_txsalmon.RData Untracked: data/covar_lm.csv Untracked: data/covar_lm_missing.csv Untracked: data/eigengenes_T1_T2_cov_adj_exp_5_modules.txt Untracked: data/eigengenes_T1_T2_module_background.txt Untracked: data/eigengenes_adj_exp_7_modules.txt Untracked: data/eigengenes_cov_adj_exp_14_modules.txt Untracked: data/eigengenes_module_background.txt Untracked: data/eigengenes_unadj_exp_10_modules.txt Untracked: data/eigengenes_unadj_exp_6_modules.txt Untracked: data/eigengenes_unadj_exp_9_modules.txt Untracked: data/files_list.txt Untracked: data/final_covariates.csv Untracked: data/gene_exp_values_2202.txt Untracked: data/gene_exp_values_2209.txt Untracked: data/gene_exp_values_2218.txt Untracked: data/gene_exp_values_2220.txt Untracked: data/gene_exp_values_2226.txt Untracked: data/gene_exp_values_2228.txt Untracked: data/gene_expression_filtered_T1T5.csv Untracked: data/gene_names_58387.txt Untracked: data/gene_to_tran.txt Untracked: data/lm_covar_fixed_random.csv Untracked: data/lm_covar_fixed_random_geno.csv Untracked: data/logs/ Untracked: data/module_T1T2_cov_adj_blue.txt Untracked: data/module_T1T2_cov_adj_brown.txt Untracked: data/module_T1T2_cov_adj_turquoise.txt Untracked: data/module_T1T2_cov_adj_yellow.txt Untracked: data/module_adj_cov_merged_blue.txt Untracked: data/module_adj_cov_merged_brown.txt Untracked: data/module_adj_cov_merged_cyan.txt Untracked: data/module_adj_cov_merged_green.txt Untracked: data/module_adj_cov_merged_greenyellow.txt Untracked: data/module_adj_cov_merged_magenta.txt Untracked: data/module_adj_cov_merged_red.txt Untracked: data/module_adj_cov_merged_salmon.txt Untracked: data/module_adj_cov_merged_tan.txt Untracked: data/module_adj_cov_merged_yellow.txt Untracked: data/module_black.txt Untracked: data/module_blue.txt Untracked: data/module_brown.txt Untracked: data/module_cov_adj_black.txt Untracked: data/module_cov_adj_blue.txt Untracked: data/module_cov_adj_brown.txt Untracked: data/module_cov_adj_cyan.txt Untracked: data/module_cov_adj_green.txt Untracked: data/module_cov_adj_greenyellow.txt Untracked: data/module_cov_adj_magenta.txt Untracked: data/module_cov_adj_pink.txt Untracked: data/module_cov_adj_purple.txt Untracked: data/module_cov_adj_red.txt Untracked: data/module_cov_adj_salmon.txt Untracked: data/module_cov_adj_tan.txt Untracked: data/module_cov_adj_turquoise.txt Untracked: data/module_cov_adj_yellow.txt Untracked: data/module_cyan.txt Untracked: data/module_green.txt Untracked: data/module_greenyellow.txt Untracked: data/module_magenta.txt Untracked: data/module_merged_black.txt Untracked: data/module_merged_blue.txt Untracked: data/module_merged_brown.txt Untracked: data/module_merged_cyan.txt Untracked: data/module_merged_green.txt Untracked: data/module_merged_greenyellow.txt Untracked: data/module_merged_magenta.txt Untracked: data/module_merged_pink.txt Untracked: data/module_merged_purple.txt Untracked: data/module_merged_red.txt Untracked: data/module_merged_salmon.txt Untracked: data/module_merged_tan.txt Untracked: data/module_merged_turquoise.txt Untracked: data/module_merged_yellow.txt Untracked: data/module_pink.txt Untracked: data/module_purple.txt Untracked: data/module_red.txt Untracked: data/module_salmon.txt Untracked: data/module_tan.txt Untracked: data/module_turquoise.txt Untracked: data/module_yellow.txt Untracked: data/notimecovariates.csv Untracked: data/only_individuals_biomarkers_weight_restoration_study.xlsx Untracked: data/pcs_genes.csv Untracked: data/pcs_genes.txt Untracked: data/rest1t2_BI_hg37.rds Untracked: data/rest1t2_BI_hg38.rds Untracked: data/rest1t2_hg37.rds Untracked: data/rest1t2_psych_meds_BMI_hg37.rds Untracked: data/rest1t2_psych_meds_hg37.rds Untracked: data/rest2t3_BI_hg37.rds Untracked: data/rest2t3_BI_hg38.rds Untracked: data/rest2t3_hg37.rds Untracked: data/rest2t3_psych_meds_BMI_hg37.rds Untracked: data/rest2t3_psych_meds_hg37.rds Untracked: data/salmon_gene_matrix_bak_reorder_time.txt Untracked: data/technical_sample_info.csv Untracked: data/tx_to_gene.txt Untracked: data/tx_to_gene_37.txt Untracked: data/usa2.pcawithref.menv.mds_cov Untracked: data/vsd_values_hg38_gc.rds Untracked: data/~$Labels.xlsx Untracked: data/~$T0_consolid.xlsx Untracked: docs/VennDiagram2018-07-24_06-55-46.log Untracked: docs/VennDiagram2018-07-24_06-56-13.log Untracked: docs/VennDiagram2018-07-24_06-56-50.log Untracked: docs/VennDiagram2018-07-24_06-58-41.log Untracked: docs/VennDiagram2018-07-24_07-00-07.log Untracked: docs/VennDiagram2018-07-24_07-00-42.log Untracked: docs/VennDiagram2018-07-24_07-01-08.log Untracked: docs/figure/ Unstaged changes: Modified: analysis/_site.yml Modified: analysis/about.Rmd Deleted: analysis/chunks.R Modified: analysis/index.Rmd Modified: analysis/license.Rmd
Introduction
# Library
library(edgeR)Loading required package: limmalibrary(limma)
library(VennDiagram)Warning: package 'VennDiagram' was built under R version 3.4.4Loading required package: gridLoading required package: futile.loggerlibrary(cowplot)Warning: package 'cowplot' was built under R version 3.4.4Loading required package: ggplot2Warning: package 'ggplot2' was built under R version 3.4.4
Attaching package: 'cowplot'The following object is masked from 'package:ggplot2':
ggsave# Read in the data
tx.salmon <- readRDS("../data/counts_hg38_gc_txsalmon.RData")
salmon_counts<- as.data.frame(tx.salmon$counts)
#tx.salmon <- readRDS("../data/counts_hg38_gc_dds.RData")
#salmon_counts<- as.data.frame(tx.salmon)
# Subset to T1-T3
salmon_counts <- salmon_counts[,1:144]
# Read in the clinical covariates
clinical_sample_info <- read.csv("../data/lm_covar_fixed_random.csv")
dim(clinical_sample_info)[1] 156 13# Subset to T1-T3
clinical_sample <- clinical_sample_info[1:144,(-12)]
dim(clinical_sample)[1] 144 12Differential expression pipeline
# Filter lowly expressed reads
cpm <- cpm(salmon_counts, log=TRUE)
expr_cutoff <- 1.5
hist(cpm, main = "log2(CPM) values in unfiltered data", breaks = 100, xlab = "log2(CPM) values")
abline(v = expr_cutoff, col = "red", lwd = 3)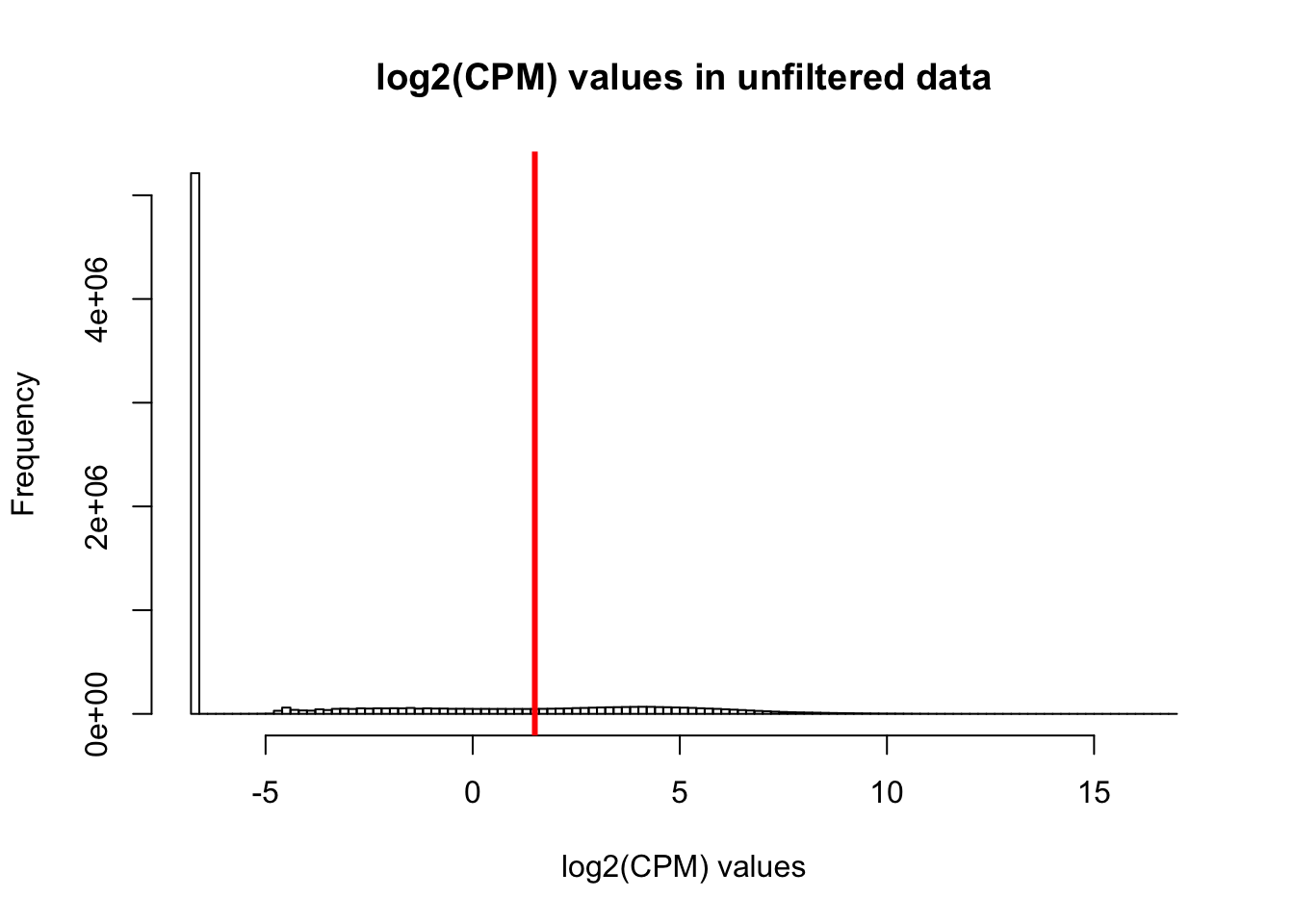
hist(cpm, main = "log2(CPM) values in unfiltered data", breaks = 100, xlab = "log2(CPM) values", ylim = c(0, 100000))
abline(v = expr_cutoff, col = "red", lwd = 3)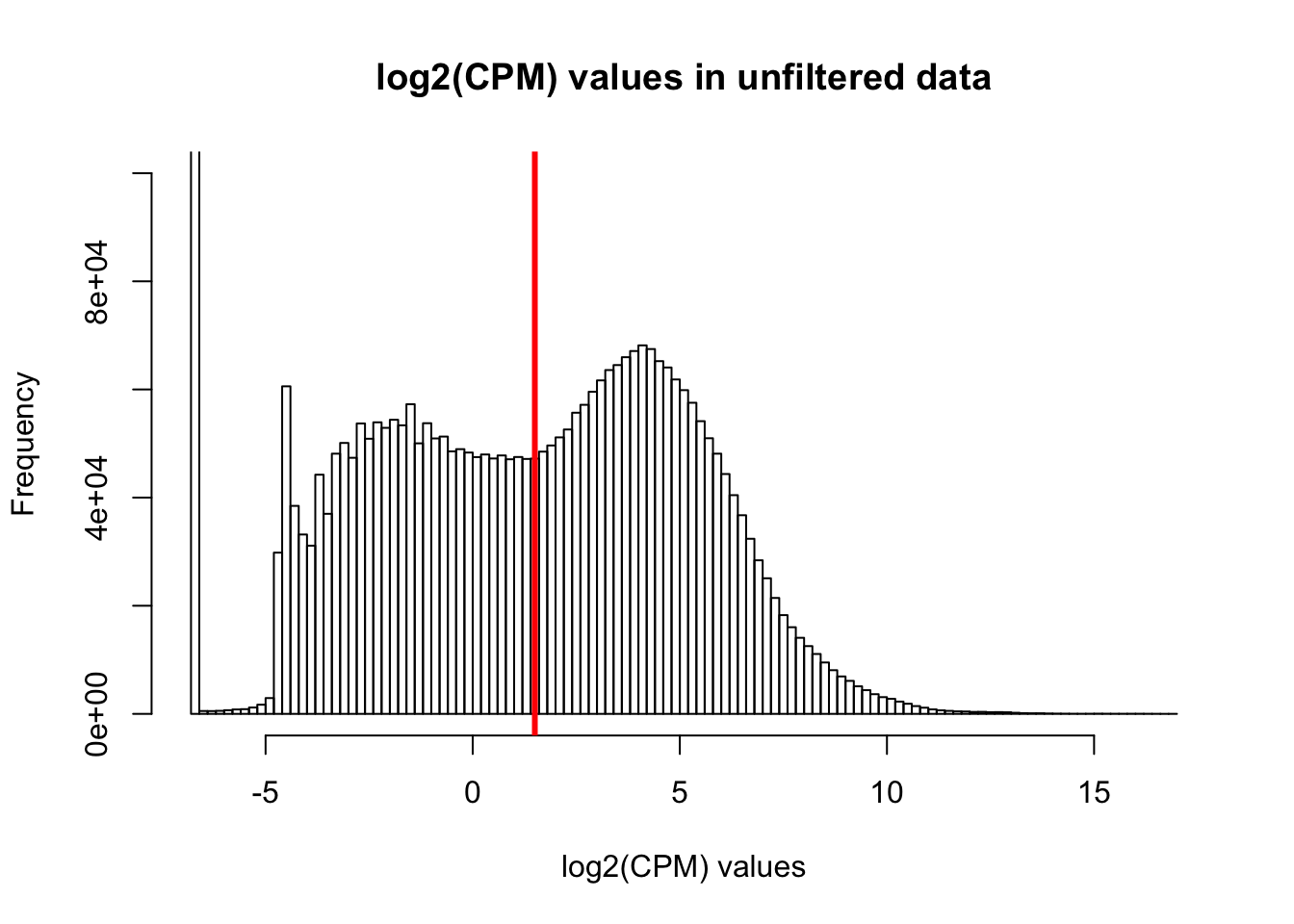
# Basic filtering
cpm_filtered <- (rowSums(cpm > 1.5) > 72)
genes_in_cutoff <- cpm[cpm_filtered==TRUE,]
hist(as.numeric(unlist(genes_in_cutoff)), main = "log2(CPM) values in filtered data", breaks = 100, xlab = "log2(CPM) values")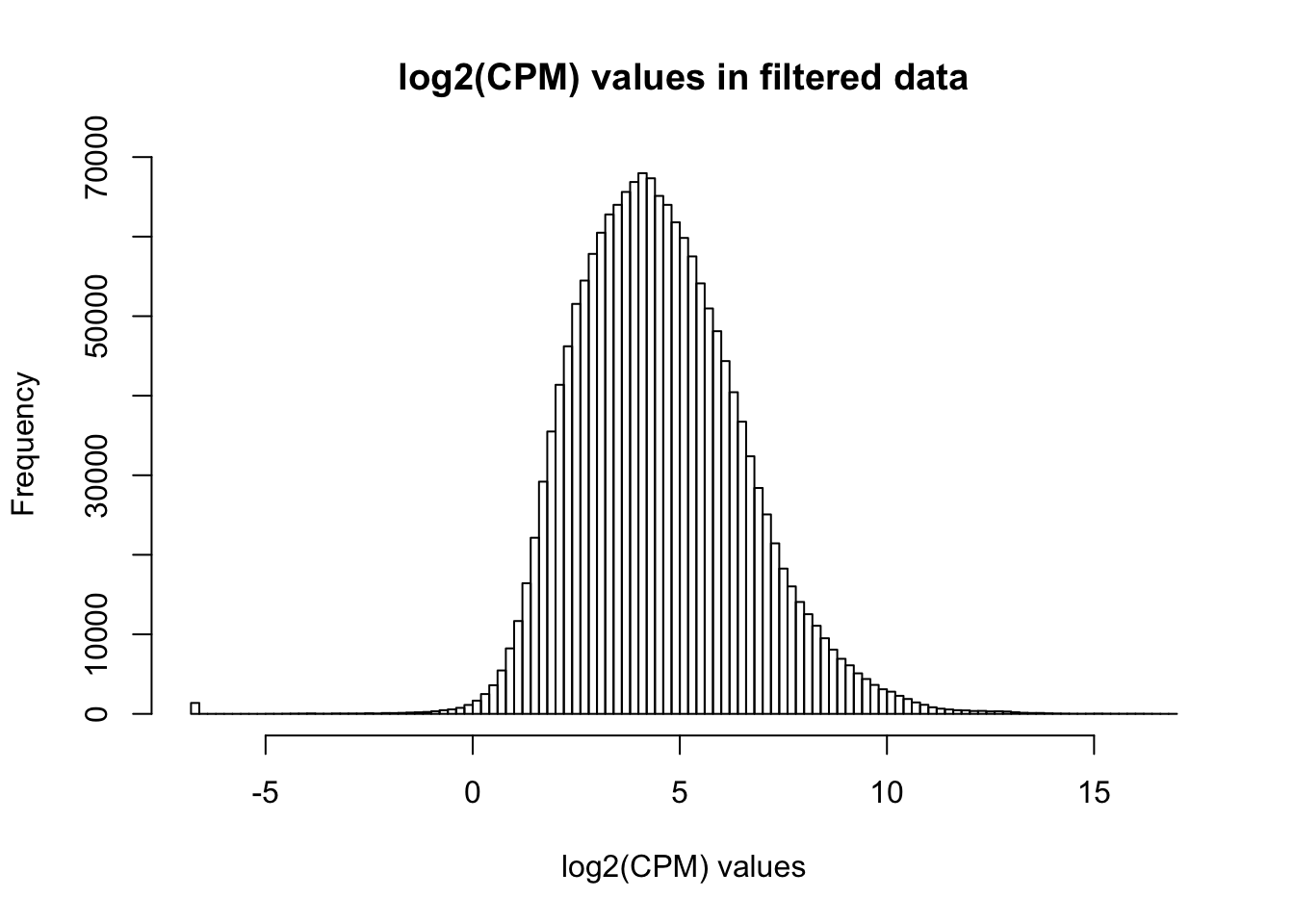
# Find the original counts of all of the genes that fit the criteria
counts_genes_in_cutoff <- salmon_counts[cpm_filtered==TRUE,]
dim(counts_genes_in_cutoff)[1] 11619 144# Filter out hemoglobin
counts_genes_in_cutoff <- counts_genes_in_cutoff[which( rownames(counts_genes_in_cutoff) != "HBB" ),]
counts_genes_in_cutoff <- counts_genes_in_cutoff[which( rownames(counts_genes_in_cutoff) != "HBA2" ),]
counts_genes_in_cutoff <- counts_genes_in_cutoff[which( rownames(counts_genes_in_cutoff) != "HBA1" ),]
# Take the TMM of the counts only for the genes that remain after filtering
dge_in_cutoff <- DGEList(counts=as.matrix(counts_genes_in_cutoff), genes=rownames(counts_genes_in_cutoff), group = as.character(t(clinical_sample$Individual)))
dge_in_cutoff <- calcNormFactors(dge_in_cutoff)
cpm_in_cutoff <- cpm(dge_in_cutoff, normalized.lib.sizes=TRUE, log=TRUE)
pca_genes <- prcomp(t(cpm_in_cutoff), scale = T, retx = TRUE, center = TRUE)
matrixpca <- pca_genes$x
PC1 <- matrixpca[,1]
PC2 <- matrixpca[,2]
pc3 <- matrixpca[,3]
pc4 <- matrixpca[,4]
pc5 <- matrixpca[,5]
pcs <- data.frame(PC1, PC2, pc3, pc4, pc5)
summary <- summary(pca_genes)
head(summary$importance[2,1:5]) PC1 PC2 PC3 PC4 PC5
0.25012 0.13000 0.08757 0.05524 0.03285 norm_count <- ggplot(data=pcs, aes(x=PC1, y=PC2, color= as.factor(clinical_sample$Time))) + geom_point(aes(colour = as.factor(clinical_sample$Time))) + ggtitle("PCA of normalized counts") + scale_color_discrete(name = "Time")
plot_grid(norm_count)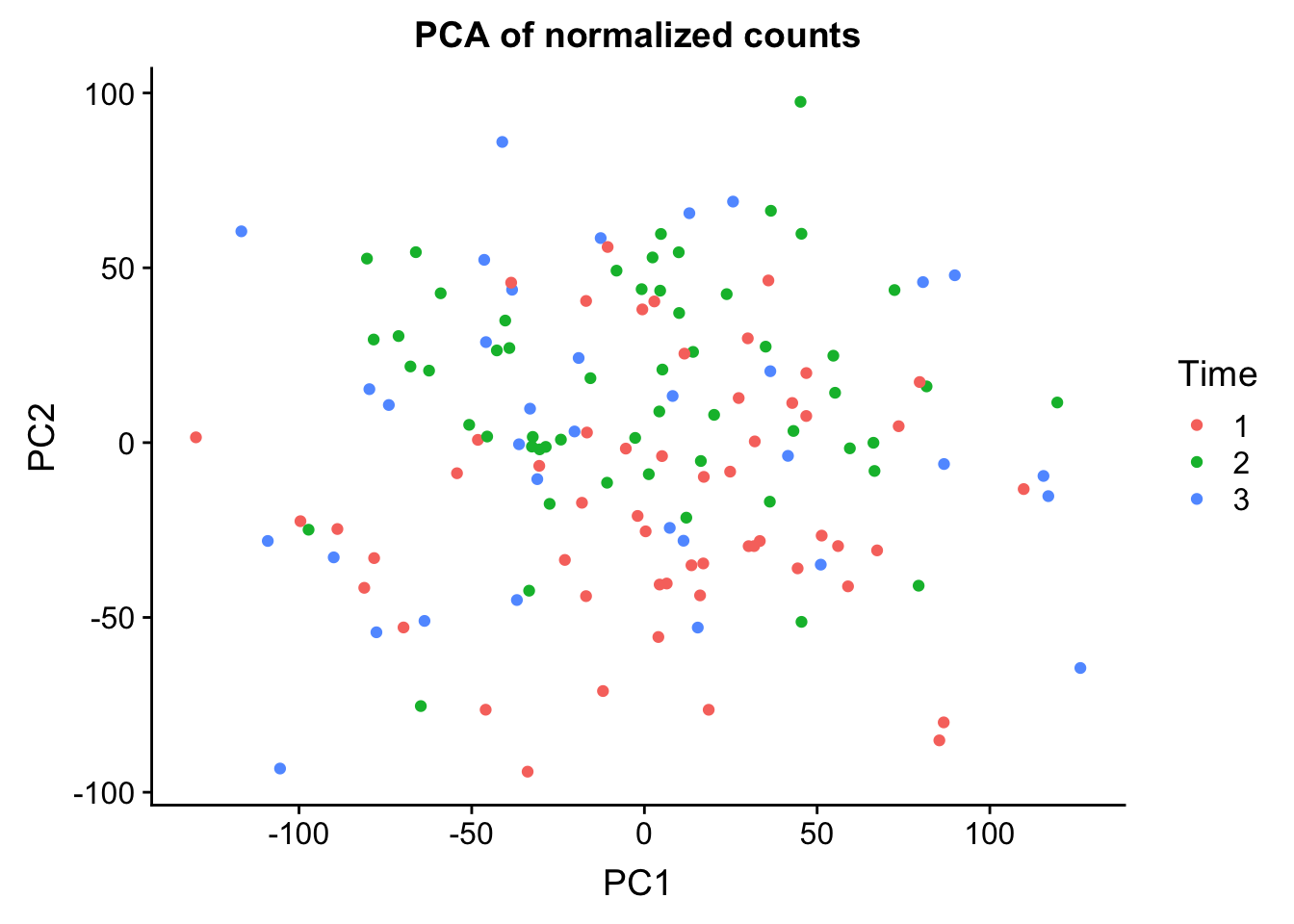
clinical_sample[,1] <- as.factor(clinical_sample[,1])
clinical_sample[,2] <- as.factor(clinical_sample[,2])
clinical_sample[,4] <- as.factor(clinical_sample[,4])
clinical_sample[,5] <- as.factor(clinical_sample[,5])
clinical_sample[,6] <- as.factor(clinical_sample[,6])
# Create the design matrix
# Use the standard treatment-contrasts parametrization. See Ch. 9 of limma
# User's Guide.
design <- model.matrix(~as.factor(Time) + Age + as.factor(Race) + as.factor(BE_GROUP) + as.factor(psychmeds) + RBC + AN + AE + AL + RIN + Change_weight, data = clinical_sample)
colnames(design) <- c("Intercept", "Time2", "Time3", "Race3", "Race5", "Age", "BE", "Psychmeds", "RBC", "AN", "AE", "AL", "RIN", "Change_weight")
# Fit model
# Model individual as a random effect.
# Recommended to run both voom and duplicateCorrelation twice.
# https://support.bioconductor.org/p/59700/#67620
cpm.voom <- voom(dge_in_cutoff, design, normalize.method="none")
#check_rel <- duplicateCorrelation(cpm.voom, design, block = clinical_sample$Individual)
check_rel_correlation <- 0.1179835
cpm.voom.corfit <- voom(dge_in_cutoff, design, normalize.method="none", plot = TRUE, block = clinical_sample$Individual, correlation = check_rel_correlation)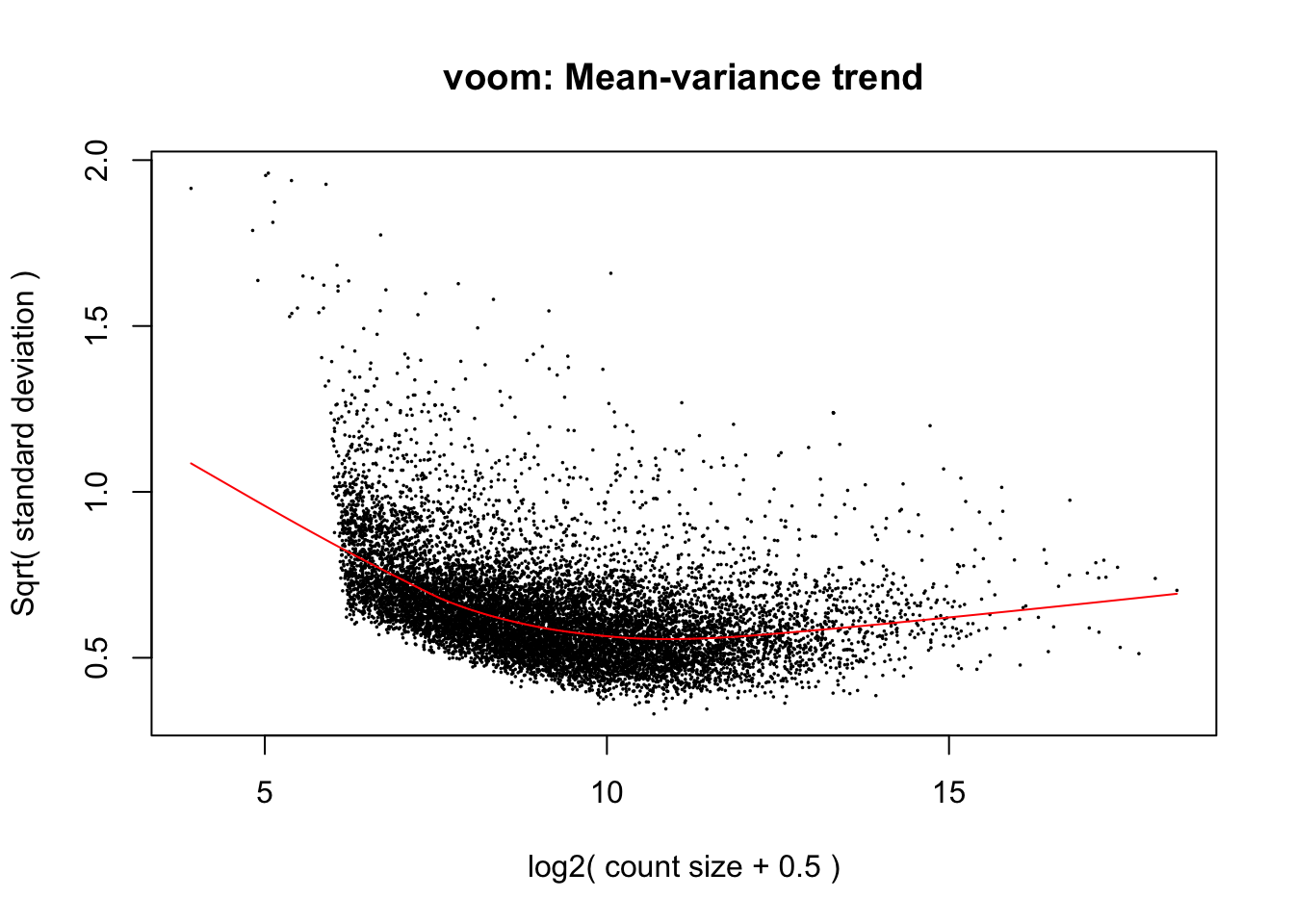
#check_rel <- duplicateCorrelation(cpm.voom.corfit, design, block = clinical_sample$Individual)
check_rel_correlation <- 0.1188083
plotDensities(cpm.voom.corfit[,1])
plotDensities(cpm.voom.corfit[,2])
plotDensities(cpm.voom.corfit[,3])
pca_genes <- prcomp(t(cpm.voom.corfit$E), scale = T, retx = TRUE, center = TRUE)
matrixpca <- pca_genes$x
PC1 <- matrixpca[,1]
PC2 <- matrixpca[,2]
pc3 <- matrixpca[,3]
pc4 <- matrixpca[,4]
pc5 <- matrixpca[,5]
pcs <- data.frame(PC1, PC2, pc3, pc4, pc5)
summary <- summary(pca_genes)
head(summary$importance[2,1:5]) PC1 PC2 PC3 PC4 PC5
0.25066 0.13028 0.08774 0.05501 0.03289 ggplot(data=pcs, aes(x=PC1, y=PC2, color=clinical_sample$Time)) + geom_point(aes(colour = as.factor(clinical_sample$Time))) + ggtitle("PCA of normalized counts") + scale_color_discrete(name = "Time")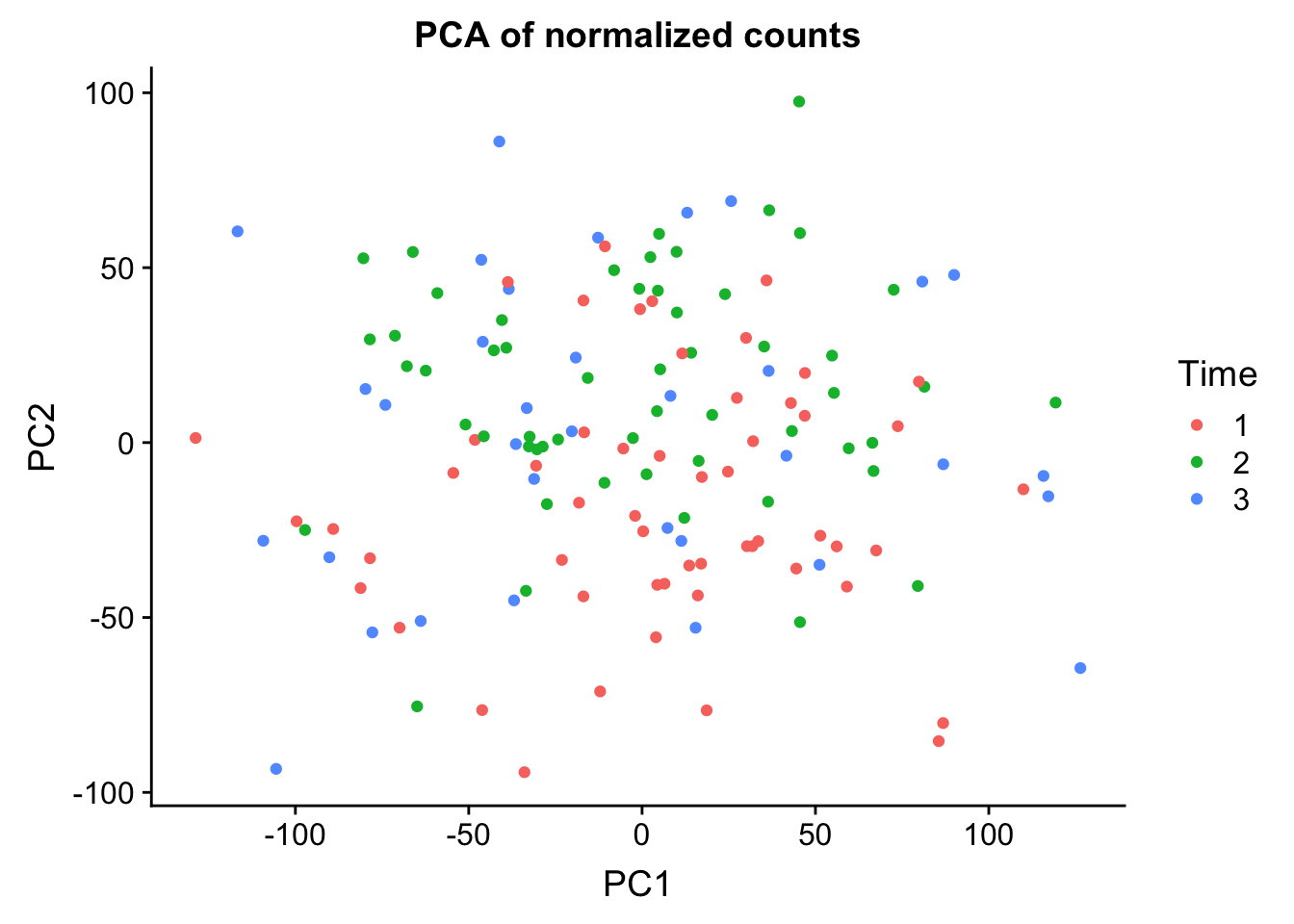
# Run lmFit and eBayes in limma
fit <- lmFit(cpm.voom.corfit, design, block=clinical_sample$Individual, correlation=check_rel_correlation)
# In the contrast matrix, have the time points
cm1 <- makeContrasts(Time1v2 = Time2, Time2v3 = Time3 - Time2, levels = design)
#cm1 <- makeContrasts(Time1v2 = Time2, Time2v3 = Time3, levels = design)
# Fit the new model
diff_species <- contrasts.fit(fit, cm1)
fit1 <- eBayes(diff_species)
FDR_level <- 0.05
Time1v2 =topTable(fit1, coef=1, adjust="BH", number=Inf, sort.by="none")
Time2v3 =topTable(fit1, coef=2, adjust="BH", number=Inf, sort.by="none")
#plot(fit1$coefficients[,1], fit1$coefficients[,2])
plot(Time1v2$logFC, Time2v3$logFC)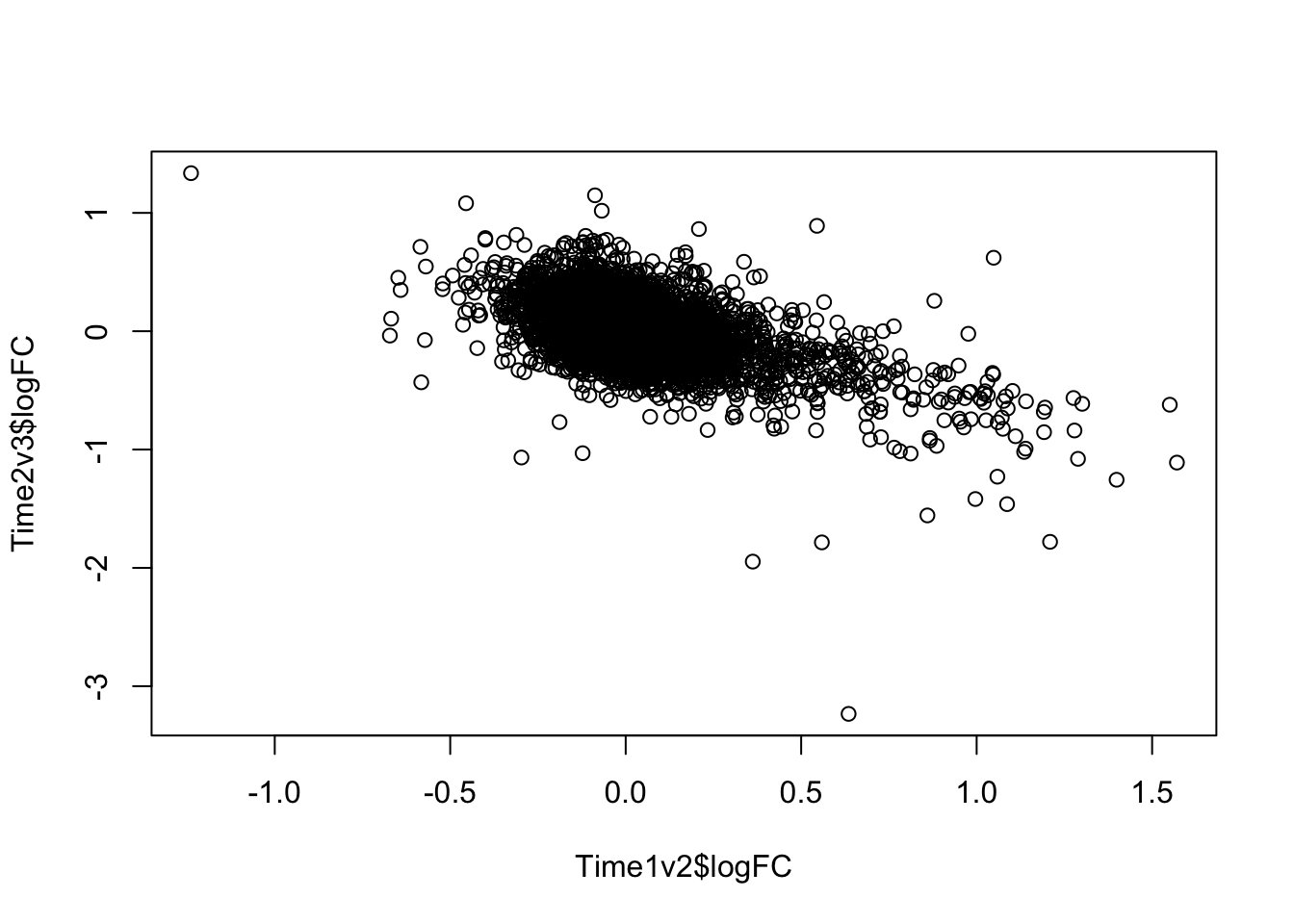
plot(Time1v2$t, Time2v3$t)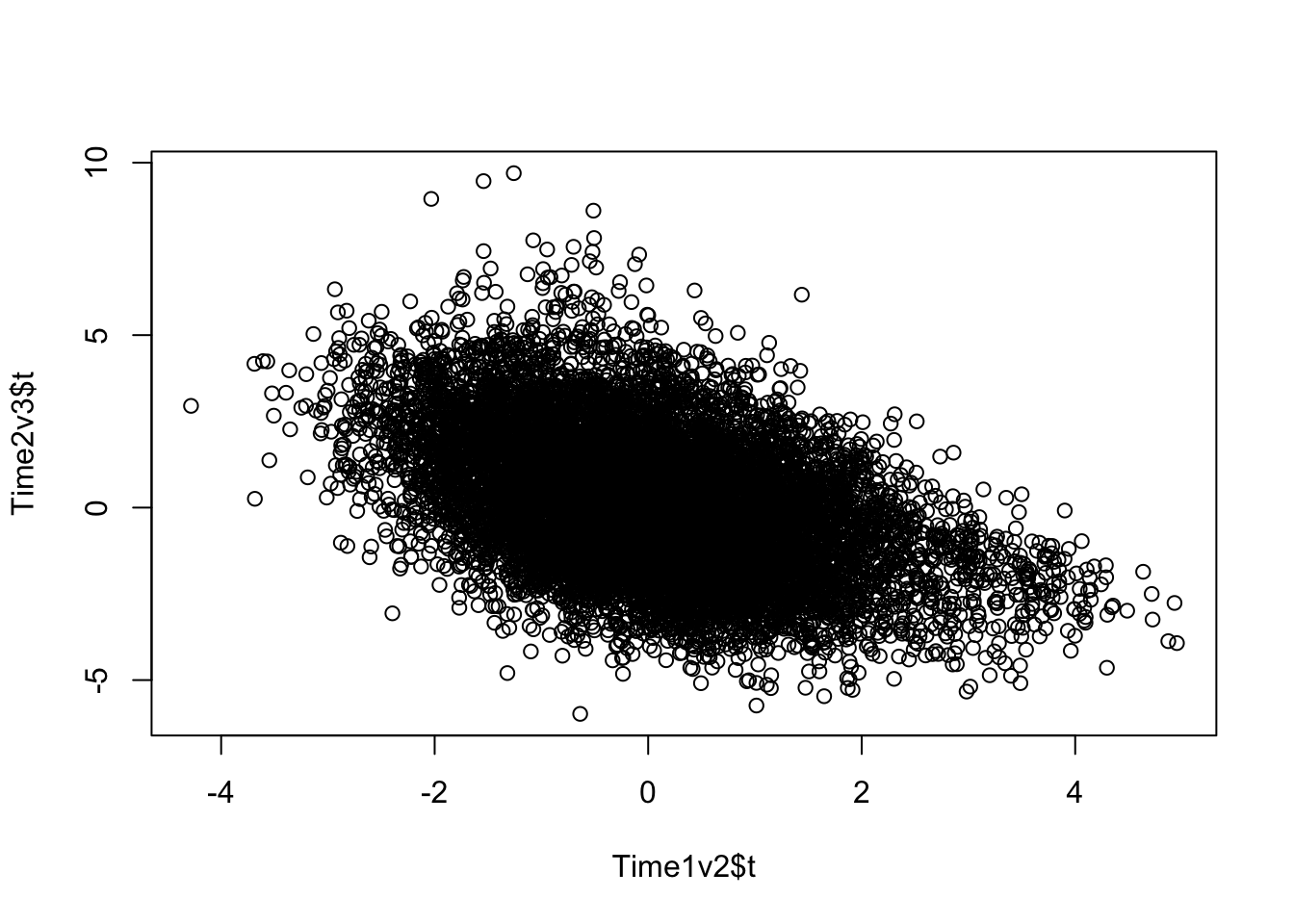
plot(Time1v2$adj.P.Val, Time2v3$adj.P.Val)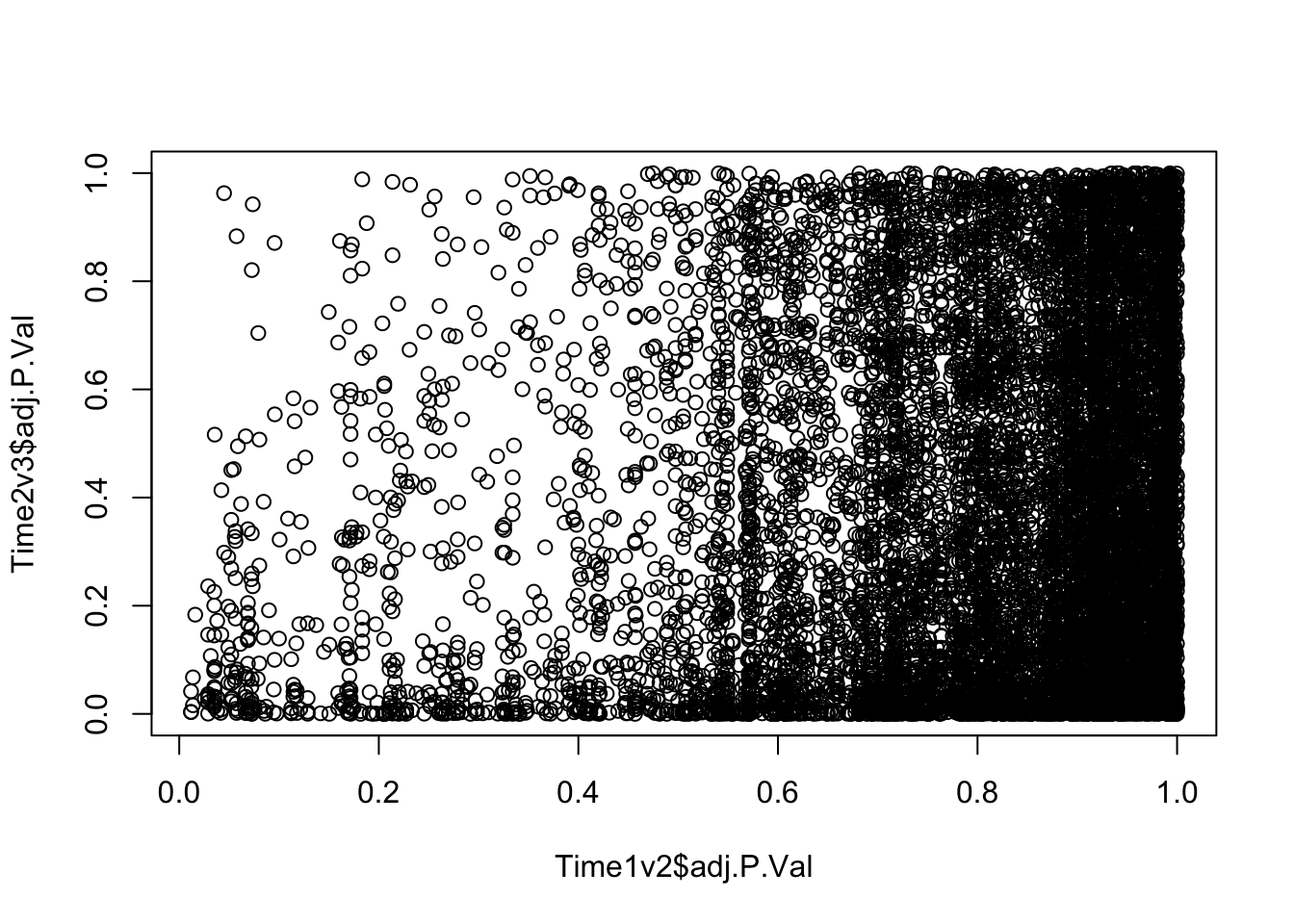
dim(Time1v2[which(Time1v2$adj.P.Val < FDR_level),])[1] 44 7dim(Time2v3[which(Time2v3$adj.P.Val < FDR_level),])[1] 1991 7head(topTable(fit1, coef=1, adjust="BH", number=100, sort.by="T")) genes logFC AveExpr t P.Value adj.P.Val
ABALON ABALON 1.2887511 2.170769 4.952850 2.150985e-06 0.01180735
CTNNAL1 CTNNAL1 1.1912596 1.961155 4.930330 2.371899e-06 0.01180735
DCAF6 DCAF6 0.5014972 5.448487 4.872156 3.049420e-06 0.01180735
RNF10 RNF10 0.8199629 8.824150 4.724856 5.712588e-06 0.01374242
GPR146 GPR146 1.1950100 4.684580 4.716587 5.915299e-06 0.01374242
ALAS2 ALAS2 1.5503665 10.196972 4.636501 8.275050e-06 0.01602050
B
ABALON 3.830754
CTNNAL1 3.499233
DCAF6 4.297388
RNF10 3.704913
GPR146 3.664174
ALAS2 3.323802head(topTable(fit1, coef=2, adjust="BH", number=100, sort.by="T")) genes logFC AveExpr t P.Value
MPHOSPH8 MPHOSPH8 0.7313353 6.149241 9.694977 3.460385e-17
CYP20A1 CYP20A1 0.6466663 4.471779 9.464323 1.306430e-16
DFFA DFFA 0.5483855 4.550302 8.948600 2.491086e-15
RTF1 RTF1 0.5960518 5.568985 8.607535 1.714095e-14
RP11-83A24.2 RP11-83A24.2 1.0179575 2.905414 7.815085 1.392854e-12
CTA-292E10.9 CTA-292E10.9 0.8056242 3.931612 7.747620 2.012435e-12
adj.P.Val B
MPHOSPH8 4.019583e-13 28.39307
CYP20A1 7.587744e-13 26.98074
DFFA 9.645486e-12 24.18296
RTF1 4.977732e-11 22.41650
RP11-83A24.2 3.235878e-09 17.81809
CTA-292E10.9 3.896074e-09 17.73477Make a Venn Diagram of the DE genes
# FDR 1%
FDR_level <- 0.01
Time12 <- rownames(Time1v2[which(Time1v2$adj.P.Val < FDR_level),])
Time23 <- rownames(Time2v3[which(Time2v3$adj.P.Val < FDR_level),])
mylist <- list()
mylist[["DE T1 to T2"]] <- Time12
mylist[["DE T2 to T3"]] <- Time23
# Make as pdf
Four_comp <- venn.diagram(mylist, filename= NULL, main="DE genes between timepoints (1% FDR)", cex=1.5 , fill = NULL, lty=1, height=2000, width=2000, rotation.degree = 180, scaled = FALSE, cat.pos = c(0,0))
grid.draw(Four_comp)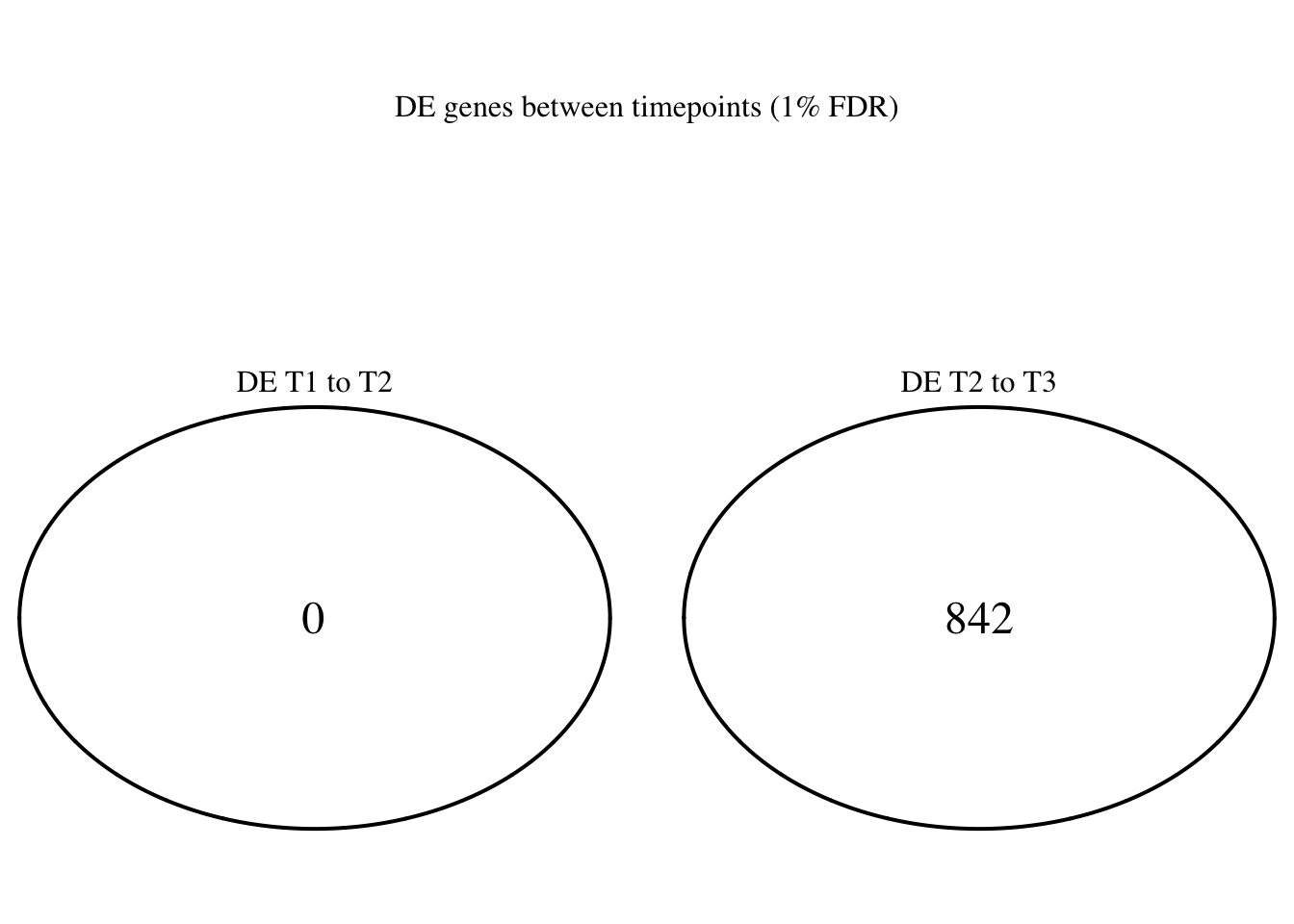
dev.off()null device
1 #pdf(file = "~/Dropbox/Figures/DET1_T2_change_weight_FDR1.pdf")
# grid.draw(Four_comp)
#dev.off()# FDR 5%
FDR_level <- 0.05
Time12 <- rownames(Time1v2[which(Time1v2$adj.P.Val < FDR_level),])
Time23 <- rownames(Time2v3[which(Time2v3$adj.P.Val < FDR_level),])
mylist <- list()
mylist[["DE T1 to T2"]] <- Time12
mylist[["DE T2 to T3"]] <- Time23
# Make as pdf
Four_comp <- venn.diagram(mylist, filename= NULL, main="DE genes between timepoints (5% FDR)", cex=1.5 , fill = NULL, lty=1, height=2000, width=2000, rotation.degree = 180, scaled = FALSE, cat.pos = c(0,0))
grid.draw(Four_comp)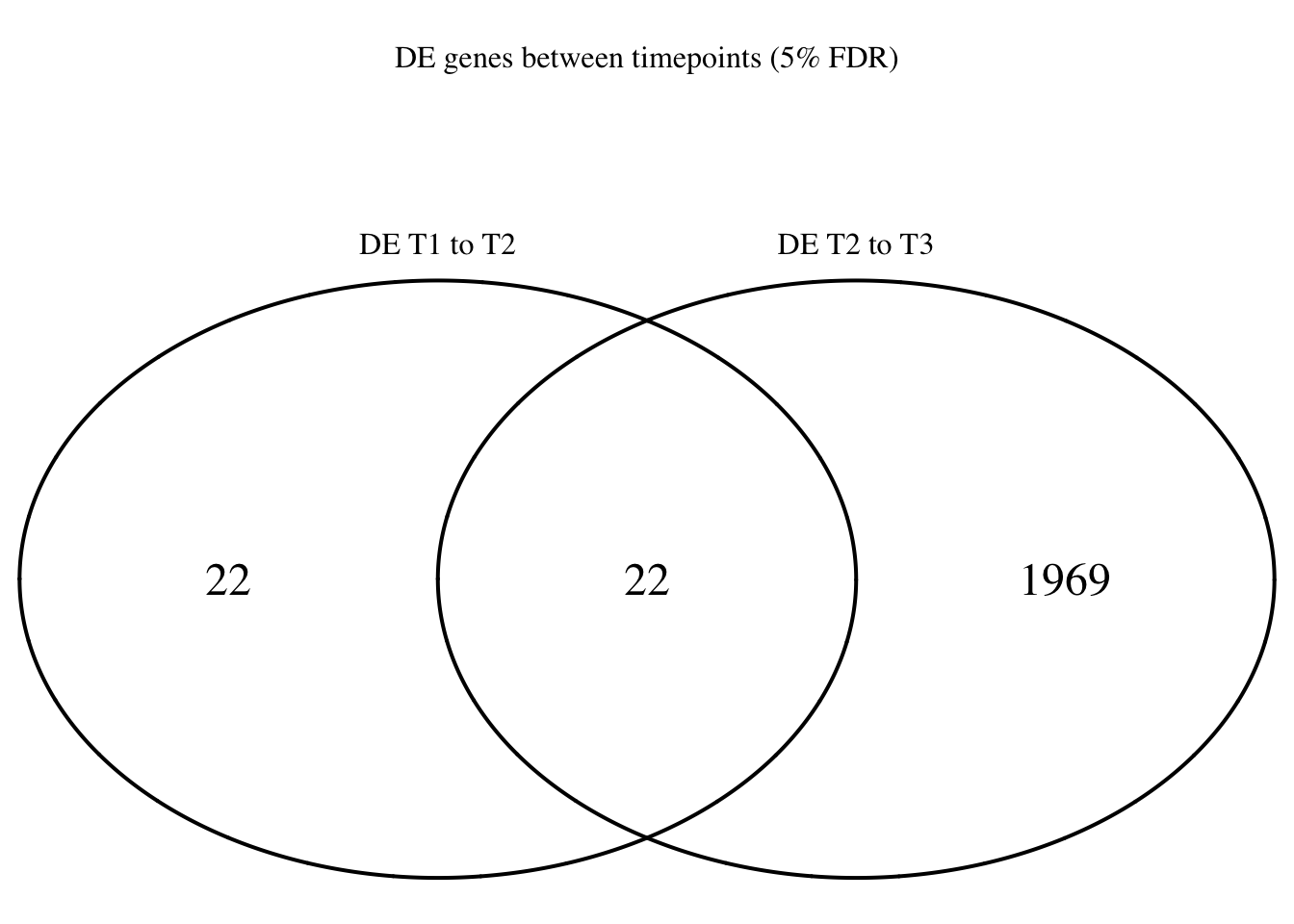
dev.off()null device
1 pdf(file = "~/Dropbox/Figures/DET1_T2_change_weight_FDR5.pdf")
grid.draw(Four_comp)
dev.off()null device
1 Using DAVID
Step 1: Use awk command to go from annotation file to ENSG and gene name only
cat gencode.v22.annotation.gtf | awk ‘BEGIN{FS=“”}{split($9,a,“;”); if($3~“gene”) print a[1]“”a[3]“”$1“:”$4“-”$5“”$7}’ | sed ‘s/gene_id “//’ | sed ’s/gene_id”//’ | sed ‘s/gene_biotype “//’| sed ’s/gene_name”//’ | sed ’s/“//g’ > Homo_sapiens.GRCh38.v22_table.txt
Step 2: Get ENSEMBL Gene IDs for background
# Set FDR level
FDR_level <- 0.01
# Get gene names after filtering
genes=as.data.frame(rownames(counts_genes_in_cutoff))
colnames(genes) <- c("Gene_name")
# Gene to ID conversion document
gene_id <- read.table("../data/Homo_sapiens.GRCh38.v22_table.txt", stringsAsFactors = FALSE)
colnames(gene_id) <- c("ENSEMBL", "Gene_name")
# Eliminate the feature after the period (for DAVID)
check <- gsub("\\..*","",gene_id$ENSEMBL)
new_gene_id <- cbind(check, gene_id$Gene_name)
gene_id <- new_gene_id
colnames(gene_id) <- c("ENSEMBL", "Gene_name")
# Get gene names of the background
comb_background <- merge(genes, gene_id, by = c("Gene_name"))
summary(duplicated(comb_background$Gene_name)) Mode FALSE TRUE
logical 11616 387 comb_background1 <- comb_background[!duplicated(comb_background$Gene_name),]
dim(comb_background1)[1] 11616 2#write.table(comb_background1$ENSEMBL, "../data/DAVID_background.txt", quote = F, row.names = F, col.names = F)Step 3: Get ENSEMBL Gene IDs for T1 to T2 list
# Get gene names after filtering
genes=as.data.frame(rownames(Time1v2[which(Time1v2$adj.P.Val < FDR_level),]))
colnames(genes) <- c("Gene_name")
# Get gene names of the list
comb_list <- merge(genes, gene_id, by = c("Gene_name"))
summary(duplicated(comb_list$Gene_name)) Mode
logical comb_list <- comb_list[!duplicated(comb_list$Gene_name),]
dim(comb_list)[1] 0 2write.table(comb_list$ENSEMBL, "../data/DAVID_list_T1T2_weight.txt", quote = F, row.names = F, col.names = F)
# Kim et al used the top 100 genes
genes=as.data.frame(rownames(topTable(fit1, coef=1, adjust="BH", number=100, sort.by="T")))
colnames(genes) <- c("Gene_name")
# Get gene names of the list
comb_list <- merge(genes, gene_id, by = c("Gene_name"))
summary(duplicated(comb_list$Gene_name)) Mode FALSE
logical 100 comb_list <- comb_list[!duplicated(comb_list$Gene_name),]
dim(comb_list)[1] 100 2write.table(comb_list$ENSEMBL, "../data/DAVID_top100_list_T1T2_weight.txt", quote = F, row.names = F, col.names = F)Step 4: Get ENSEMBL Gene IDs for T2 to T3 list
# Get gene names after filtering
genes=as.data.frame(rownames(Time2v3[which(Time2v3$adj.P.Val < FDR_level),]))
colnames(genes) <- c("Gene_name")
# Get gene names of the list
comb_list <- merge(genes, gene_id, by = c("Gene_name"))
summary(duplicated(comb_list$Gene_name)) Mode FALSE TRUE
logical 842 6 comb_list <- comb_list[!duplicated(comb_list$Gene_name),]
dim(comb_list)[1] 842 2write.table(comb_list$ENSEMBL, "../data/DAVID_list_T2T3_weight.txt", quote = F, row.names = F, col.names = F)
# Kim et al used the top 100 genes
genes=as.data.frame(rownames(topTable(fit1, coef=2, adjust="BH", number=100, sort.by="T")))
colnames(genes) <- c("Gene_name")
# Get gene names of the list
comb_list <- merge(genes, gene_id, by = c("Gene_name"))
summary(duplicated(comb_list$Gene_name)) Mode FALSE TRUE
logical 100 1 comb_list <- comb_list[!duplicated(comb_list$Gene_name),]
dim(comb_list)[1] 100 2write.table(comb_list$ENSEMBL, "../data/DAVID_top100_list_T2T3_weight.txt", quote = F, row.names = F, col.names = F)Step 5: Upload or copy and paste the gene lists to DAVID at https://david.ncifcrf.gov/conversion.jsp. Note, larger gene lists are more likely to need to be copied and pasted in the “Upload” tab, rather than uploaded.
Session information
sessionInfo()R version 3.4.3 (2017-11-30)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: OS X El Capitan 10.11.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.4/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] grid stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] cowplot_0.9.3 ggplot2_3.0.0 VennDiagram_1.6.20
[4] futile.logger_1.4.3 edgeR_3.20.9 limma_3.34.9
loaded via a namespace (and not attached):
[1] Rcpp_0.12.18 bindr_0.1.1 compiler_3.4.3
[4] pillar_1.3.0 formatR_1.5 git2r_0.23.0
[7] plyr_1.8.4 workflowr_1.1.1 R.methodsS3_1.7.1
[10] futile.options_1.0.1 R.utils_2.6.0 tools_3.4.3
[13] digest_0.6.16 evaluate_0.11 tibble_1.4.2
[16] gtable_0.2.0 lattice_0.20-35 pkgconfig_2.0.2
[19] rlang_0.2.2 yaml_2.2.0 bindrcpp_0.2.2
[22] withr_2.1.2 stringr_1.3.1 dplyr_0.7.6
[25] knitr_1.20 tidyselect_0.2.4 locfit_1.5-9.1
[28] rprojroot_1.3-2 glue_1.3.0 R6_2.2.2
[31] rmarkdown_1.10 purrr_0.2.5 lambda.r_1.2.3
[34] magrittr_1.5 whisker_0.3-2 backports_1.1.2
[37] scales_1.0.0 htmltools_0.3.6 assertthat_0.2.0
[40] colorspace_1.3-2 labeling_0.3 stringi_1.2.4
[43] lazyeval_0.2.1 munsell_0.5.0 crayon_1.3.4
[46] R.oo_1.22.0
This reproducible R Markdown analysis was created with workflowr 1.1.1